library("seriation")
data("iris")
x <- as.matrix(iris[sample(nrow(iris)), -5])
d <- dist(x)A Comparison of Seriation Methods
Introduction
This document compares the seriation methods available in package using the popular Iris data set and randomize the order of the objects.
Preparing the data
Distance seriation
Methods
We first register more seriation methods. Some of these methods require the installation of more packages.
register_DendSer()
register_optics()
register_smacof()The following methods will be used (a few slow methods are skipped).
methods <- sort(list_seriation_methods("dist"))
# skip slow methods
methods <- setdiff(methods, c("BBURCG", "BBWRCG", "Enumerate", "GSA"))
methods [1] "ARSA" "DendSer" "DendSer_ARc" "DendSer_BAR"
[5] "DendSer_LPL" "DendSer_PL" "GW" "GW_average"
[9] "GW_complete" "GW_single" "GW_ward" "HC"
[13] "HC_average" "HC_complete" "HC_single" "HC_ward"
[17] "Identity" "isomap" "isoMDS" "MDS"
[21] "MDS_angle" "MDS_smacof" "metaMDS" "monoMDS"
[25] "OLO" "OLO_average" "OLO_complete" "OLO_single"
[29] "OLO_ward" "optics" "QAP_2SUM" "QAP_BAR"
[33] "QAP_Inertia" "QAP_LS" "R2E" "Random"
[37] "Reverse" "Sammon_mapping" "SGD" "Spectral"
[41] "Spectral_norm" "SPIN_NH" "SPIN_STS" "TSP"
[45] "VAT" Details about the method can be found in the manual page for seriate().
Performing seriation
We use a loop to run the function seriate() with each method and calculate criterion measures which indicate how good the order is.
orders <- list()
criterion <- list()
for (m in methods) {
cat(m)
tm <- system.time(orders[[m]] <- seriate(d, method = m))
criterion[[m]] <- data.frame(time = tm[1]+tm[2], rbind(criterion(d, orders[[m]])))
cat(" took", tm[1]+tm[2], "sec.\n")
}ARSA took 0.776 sec.
DendSer took 0.728 sec.
DendSer_ARc took 1.014 sec.
DendSer_BAR took 0.816 sec.
DendSer_LPL took 1.057 sec.
DendSer_PL took 0.914 sec.
GW took 0.107 sec.
GW_average took 0.113 sec.
GW_complete took 0.102 sec.
GW_single took 0.108 sec.
GW_ward took 0.113 sec.
HC took 0.113 sec.
HC_average took 0.114 sec.
HC_complete took 0.113 sec.
HC_single took 0.114 sec.
HC_ward took 0.117 sec.
Identity took 0.108 sec.
isomapRegistered S3 method overwritten by 'vegan':
method from
reorder.hclust gclus took 0.144 sec.
isoMDS took 0.14 sec.
MDS took 0.108 sec.
MDS_angle took 0.117 sec.
MDS_smacof took 0.12 sec.
metaMDS took 0.428 sec.
monoMDS took 0.116 sec.
OLO took 0.103 sec.
OLO_average took 0.104 sec.
OLO_complete took 0.101 sec.
OLO_single took 0.104 sec.
OLO_ward took 0.105 sec.
optics took 0.11 sec.
QAP_2SUM took 0.139 sec.
QAP_BAR took 0.119 sec.
QAP_Inertia took 0.118 sec.
QAP_LS took 0.118 sec.
R2E took 0.118 sec.
Random took 0.103 sec.
Reverse took 0.104 sec.
Sammon_mapping took 0.111 sec.
SGD took 48.63 sec.
Spectral took 0.11 sec.
Spectral_norm took 0.119 sec.
SPIN_NH took 0.424 sec.
SPIN_STS took 0.534 sec.
TSP took 0.114 sec.
VAT took 0.109 sec.criterion <- do.call(rbind, criterion)We align the seriation orders. The reason is that an order 1, 2, 3 and 3, 2, 1 are equivalent and just an artifact of the algorithm. Aligning will reverse some orders so they are in roughly the same order. Then we sort the orders from best to worst according to a popular seriation criterion measure called Gradient_weighted.
orders <- ser_align(orders)
best_to_worse <- order(criterion[["Gradient_weighted"]], decreasing = TRUE)
orders <- orders[best_to_worse]
criterion <- criterion[best_to_worse, ]Visualize the results
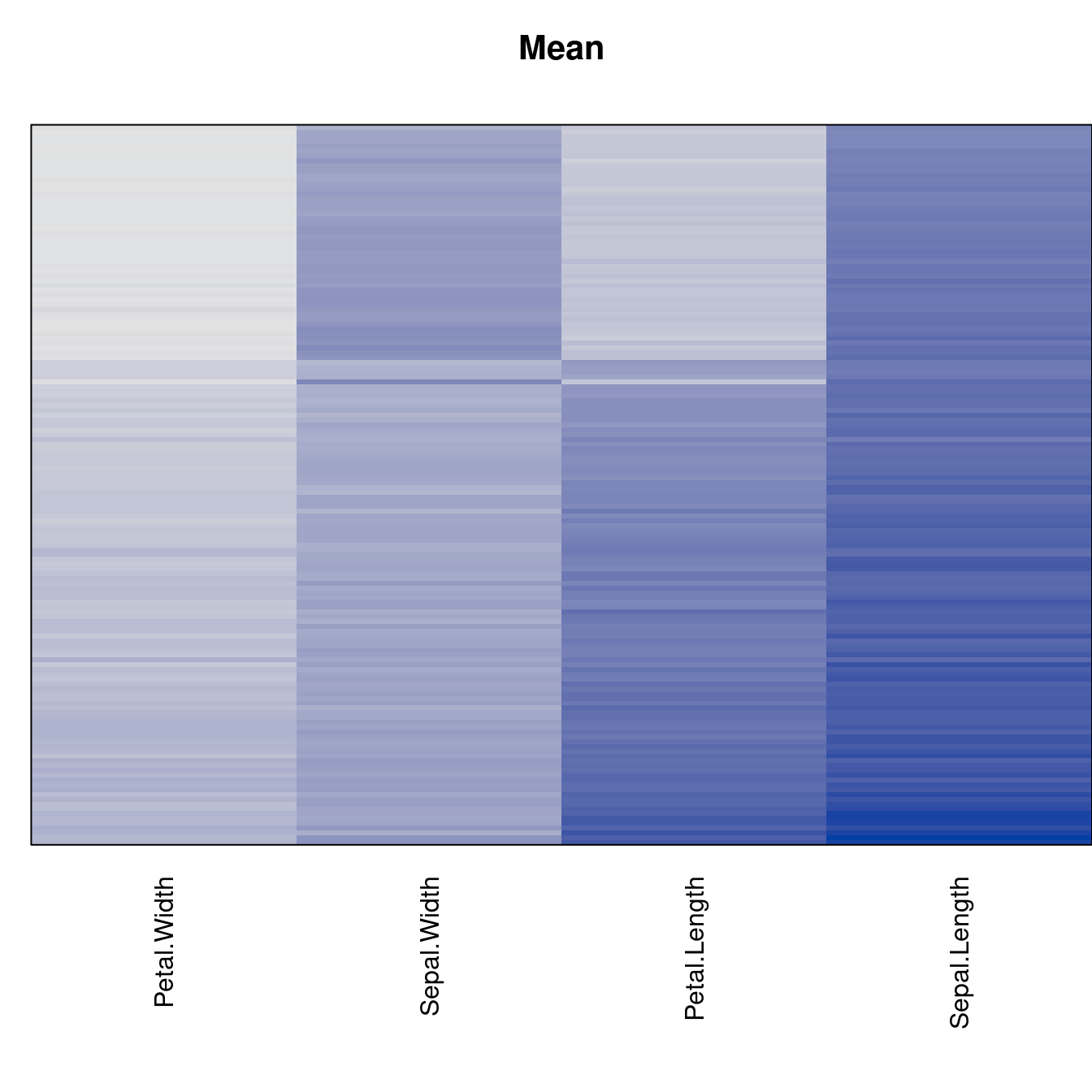
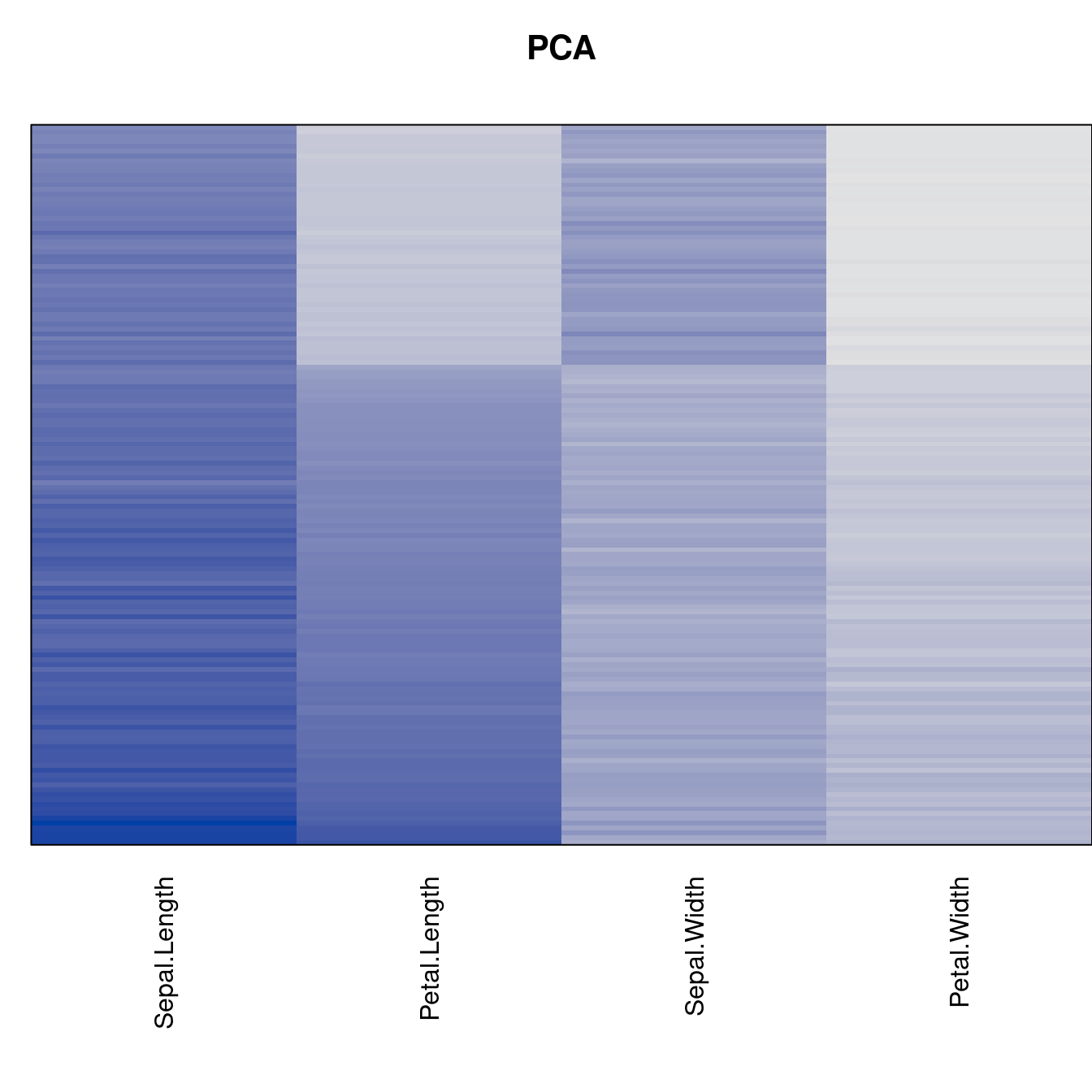
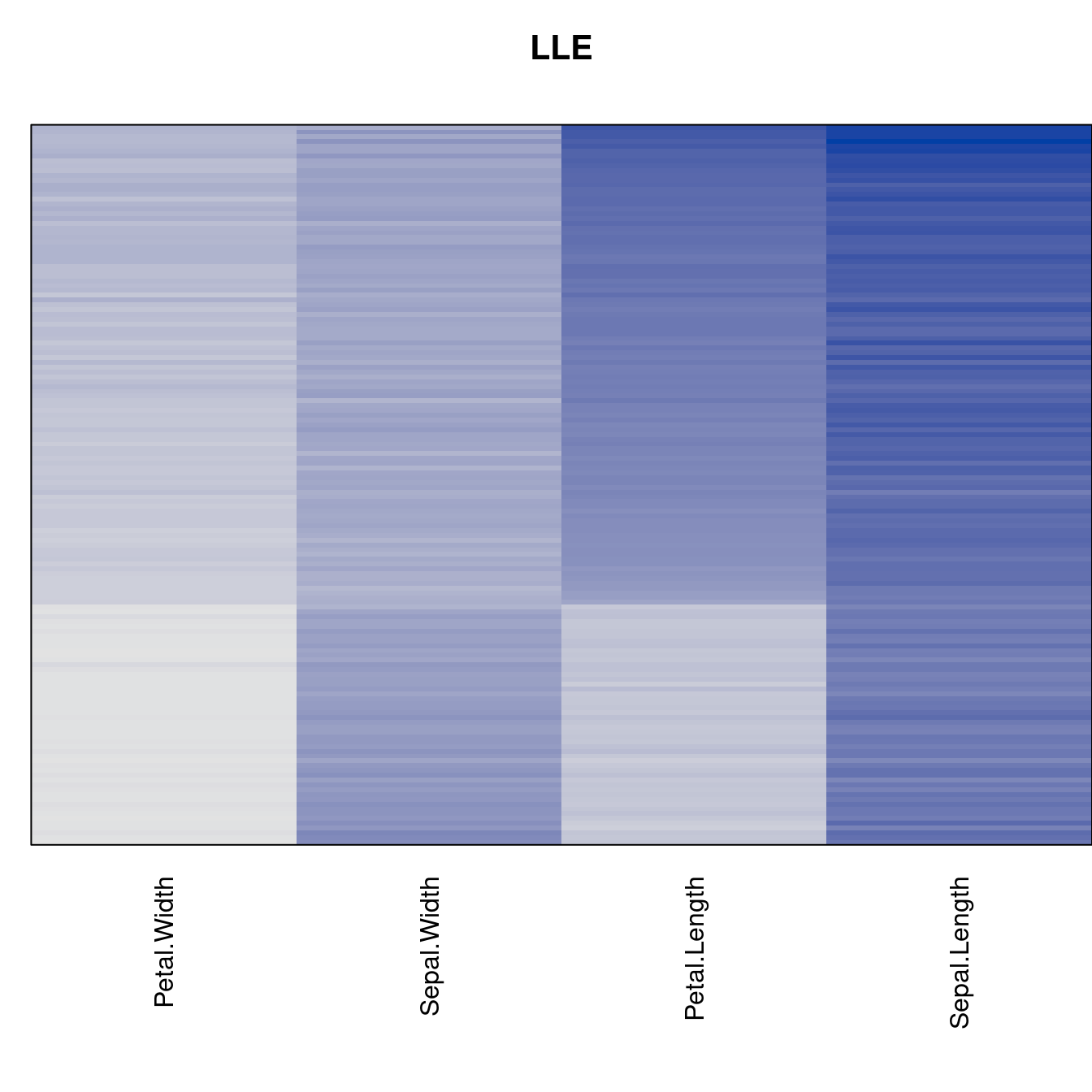
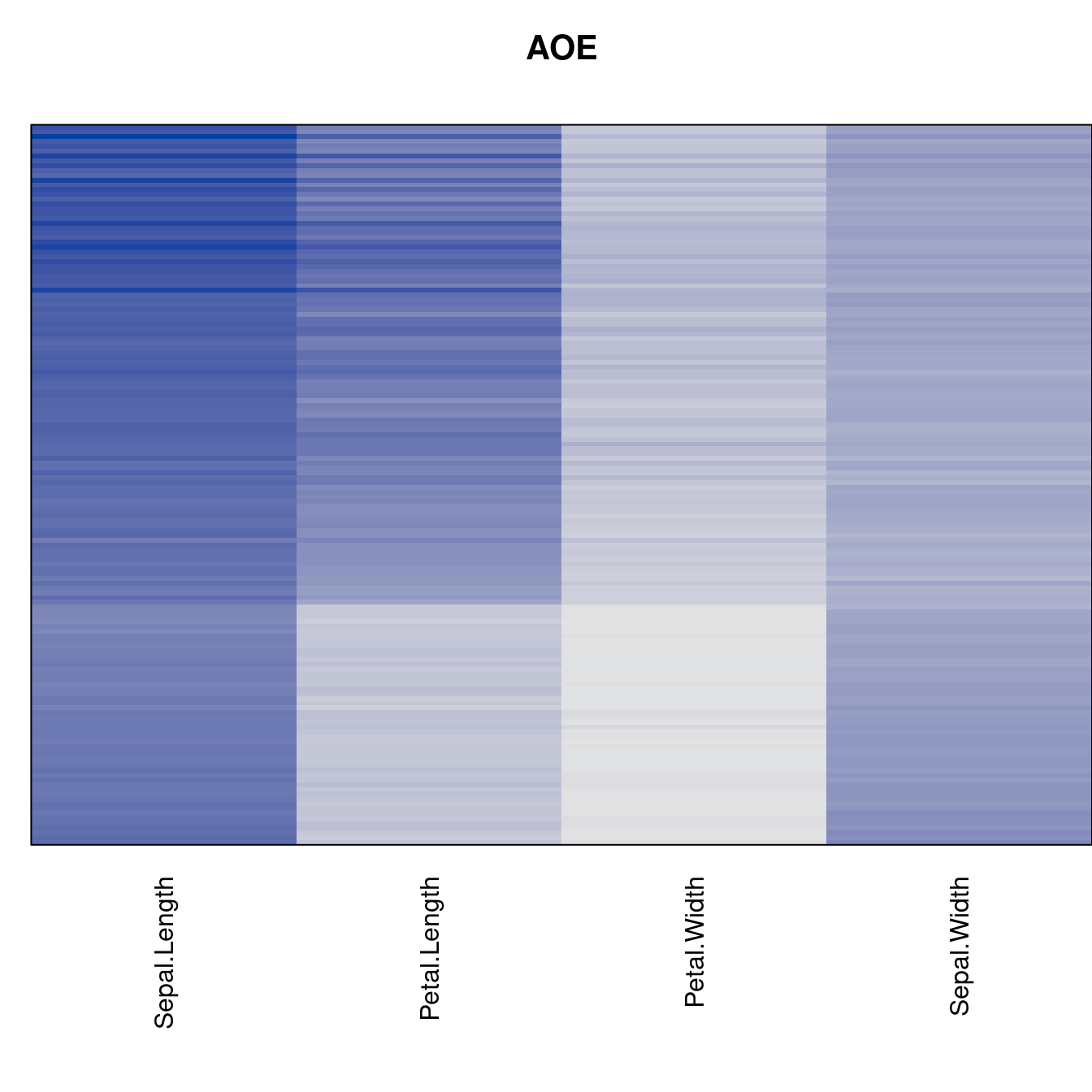
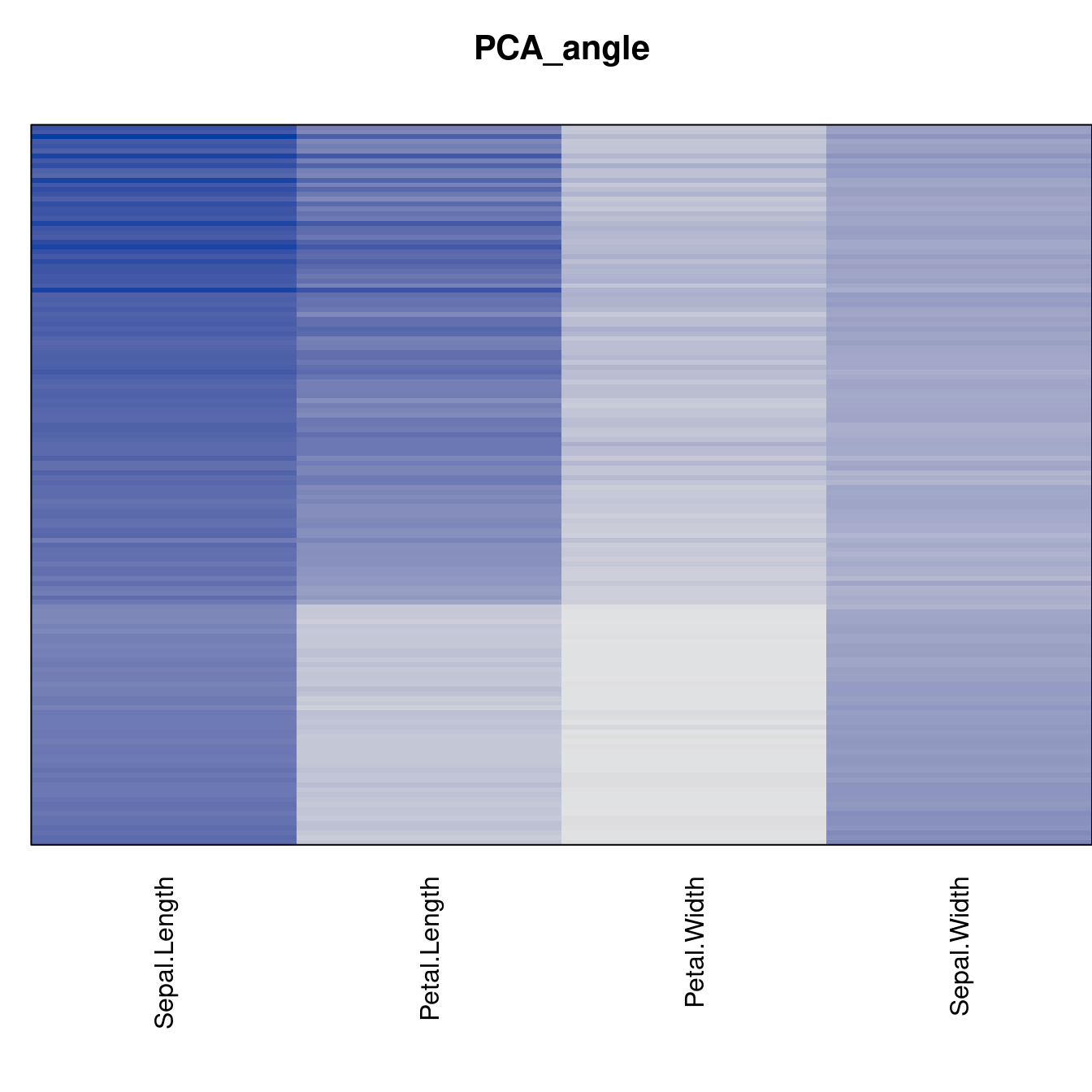
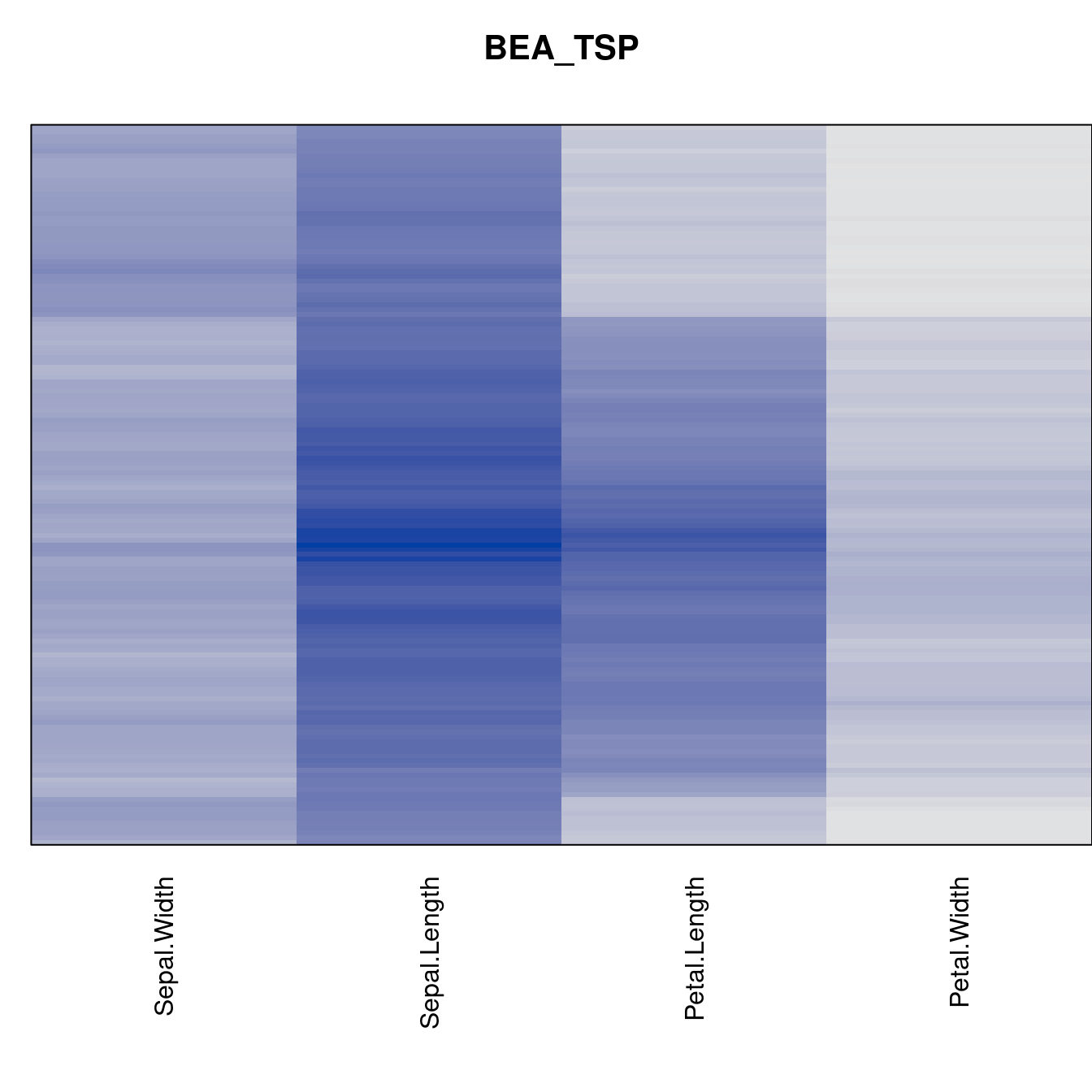
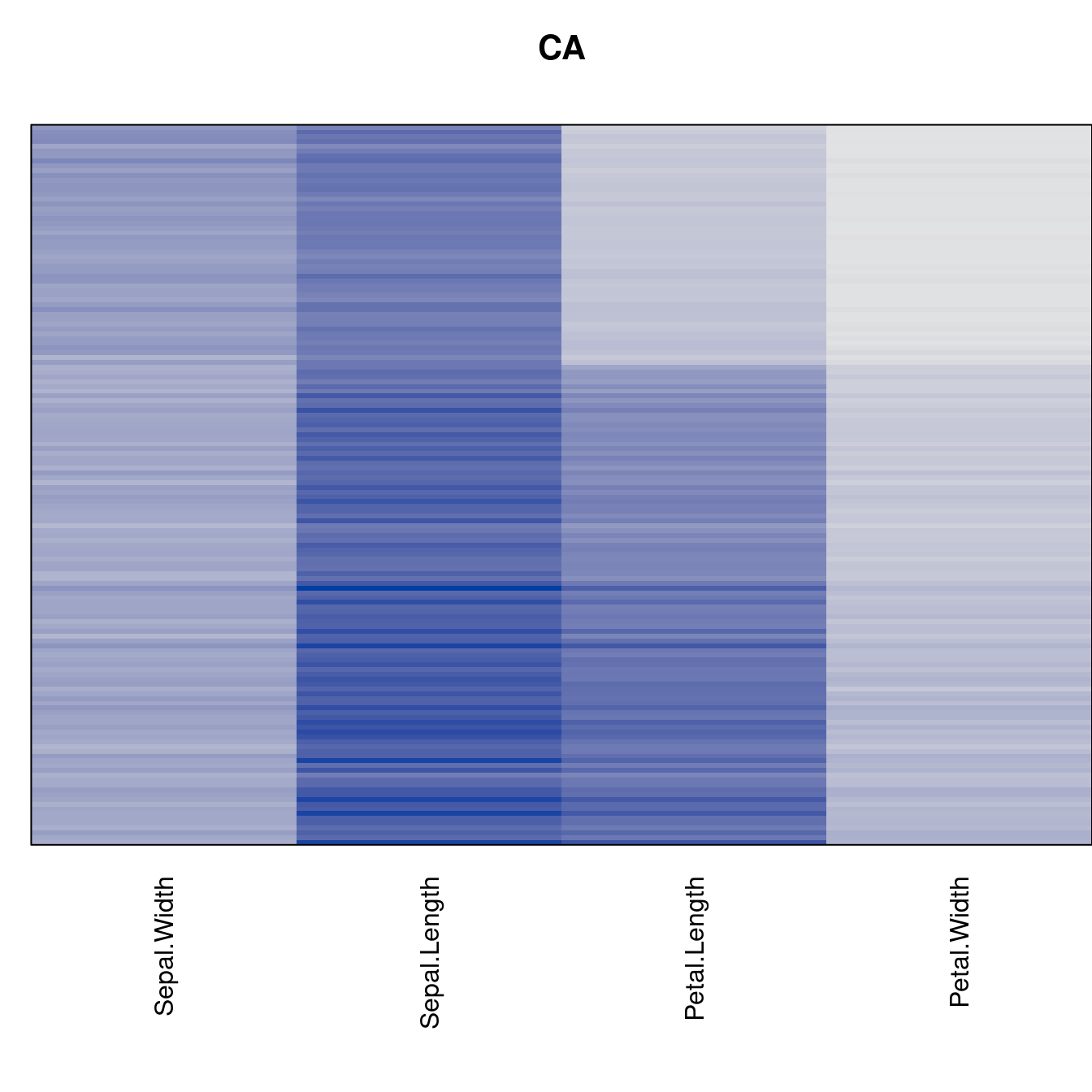
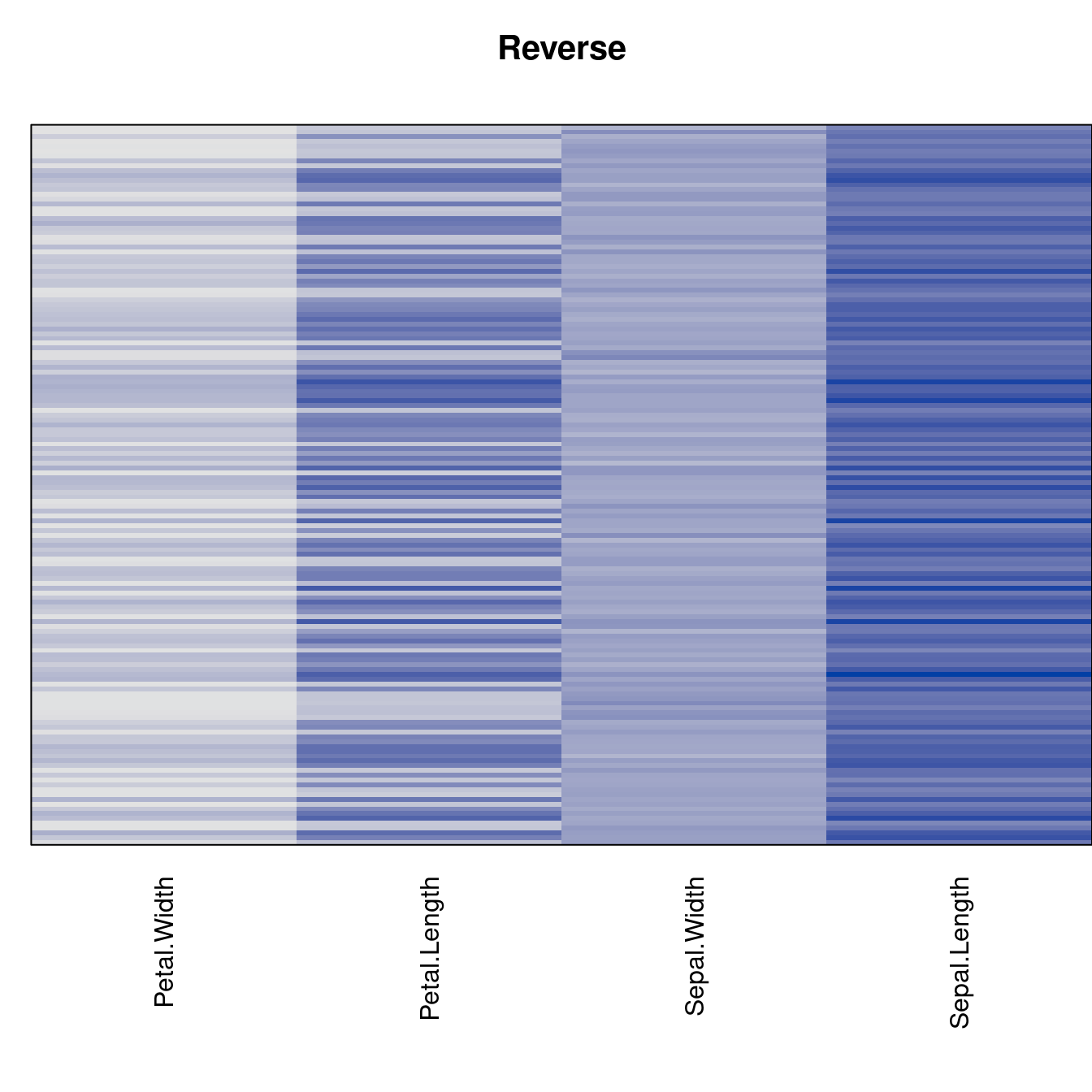
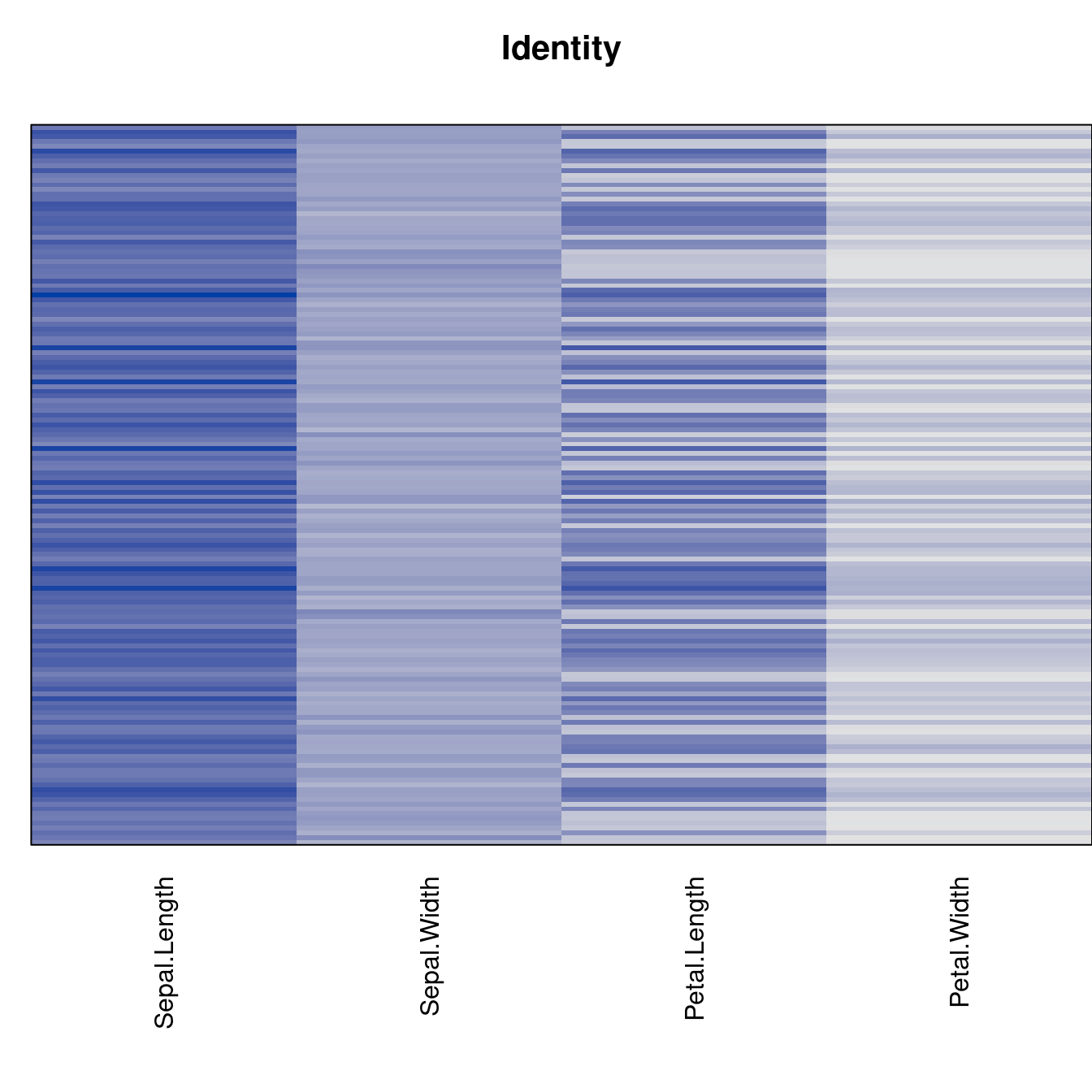
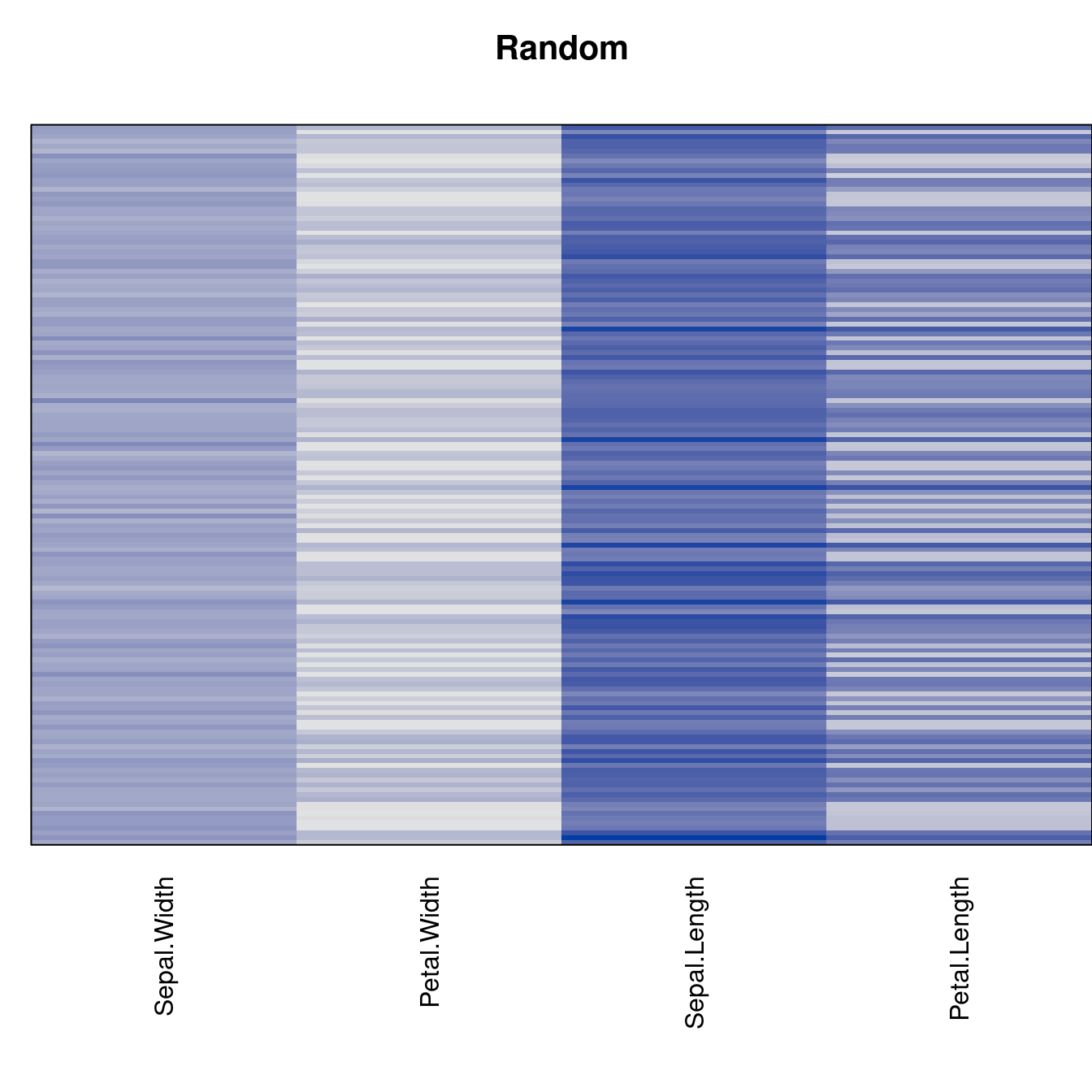
Plot the reordered dissimilarity matrices. Dark blocks along the main diagonal mean that the order reveals a “cluster” of similar objects. The Iris dataset contains three species, but two of them are very similar, so we expect to see one smaller block and one larger block.
for (n in names(orders))
pimage(d, orders[[n]], main = n , key = FALSE)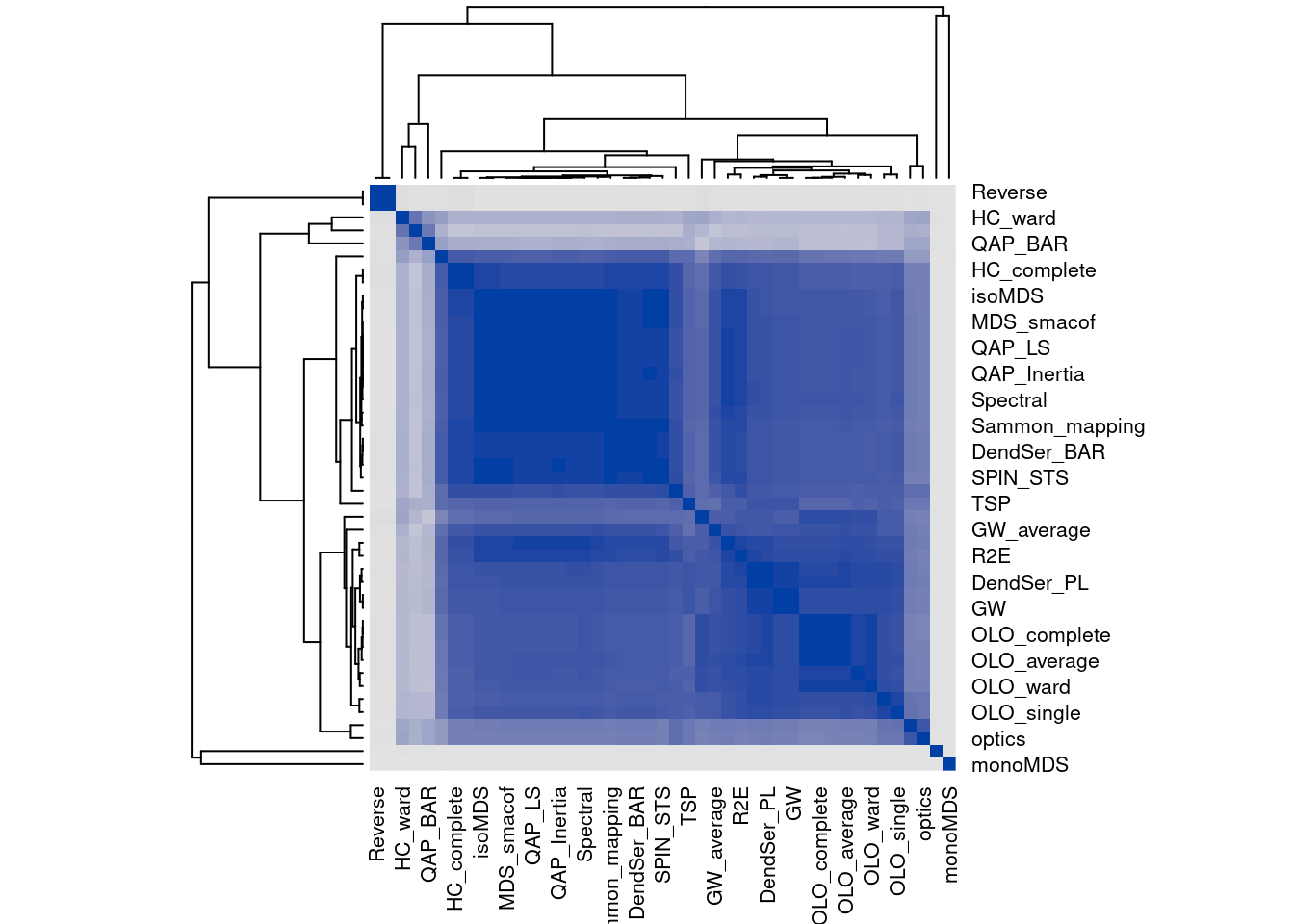
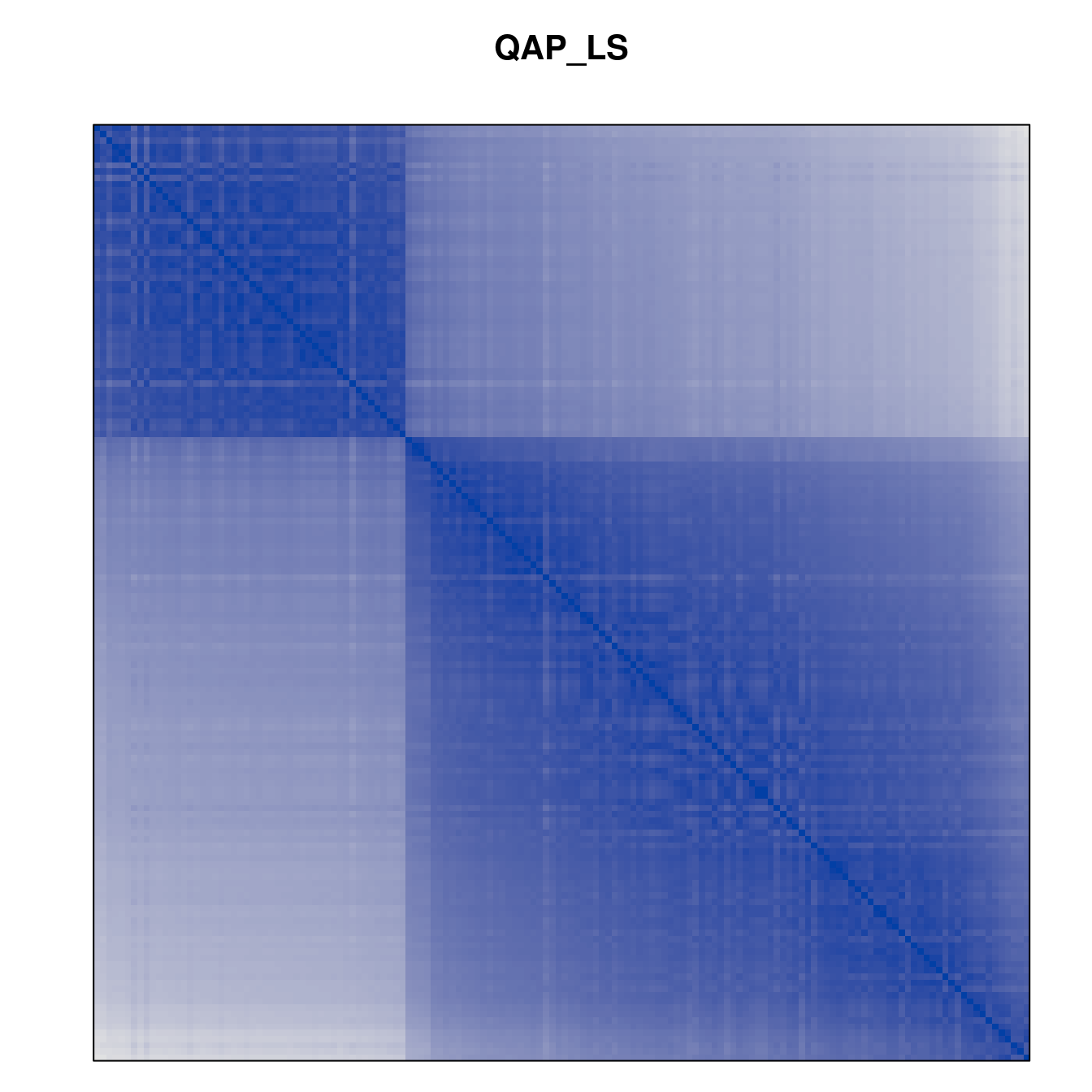
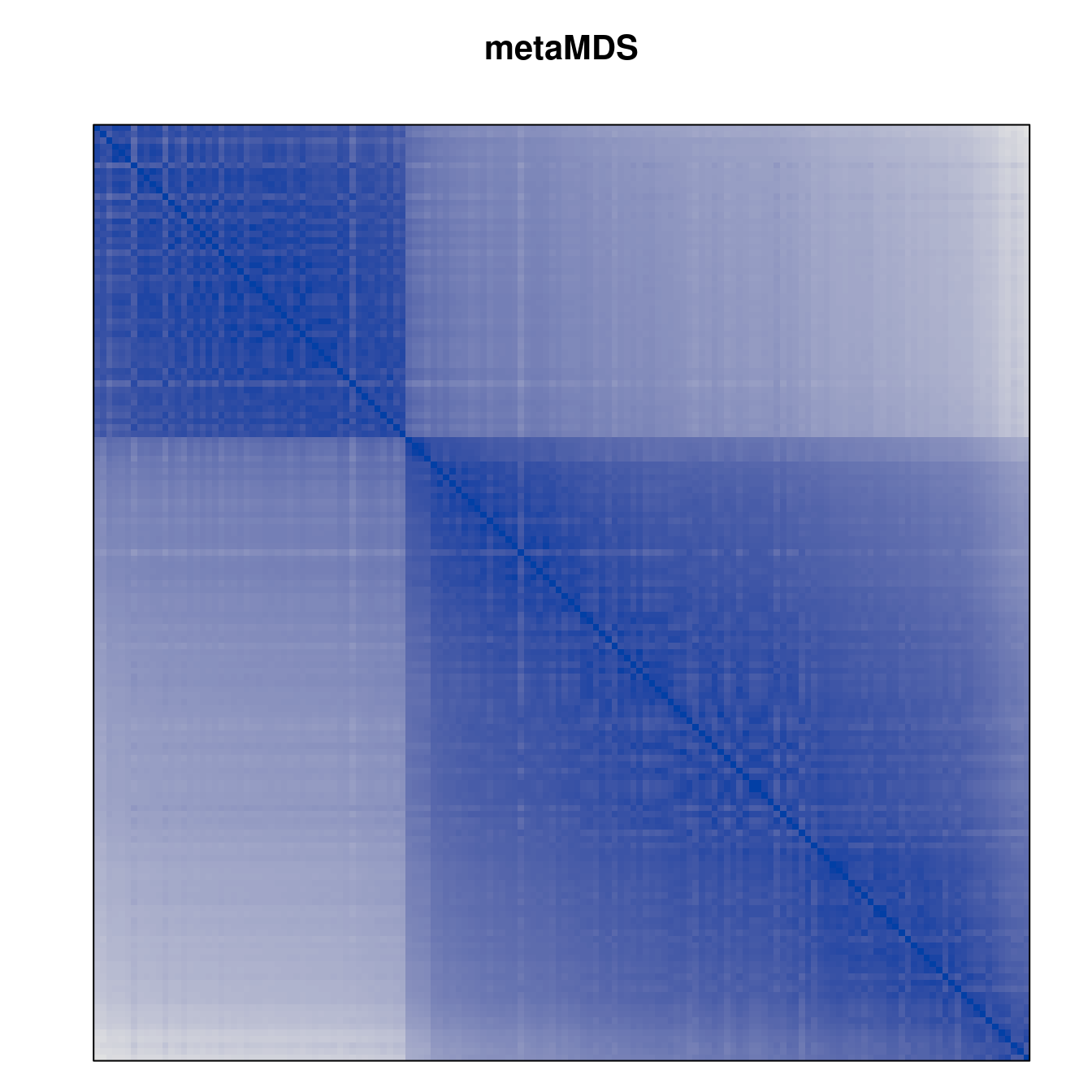
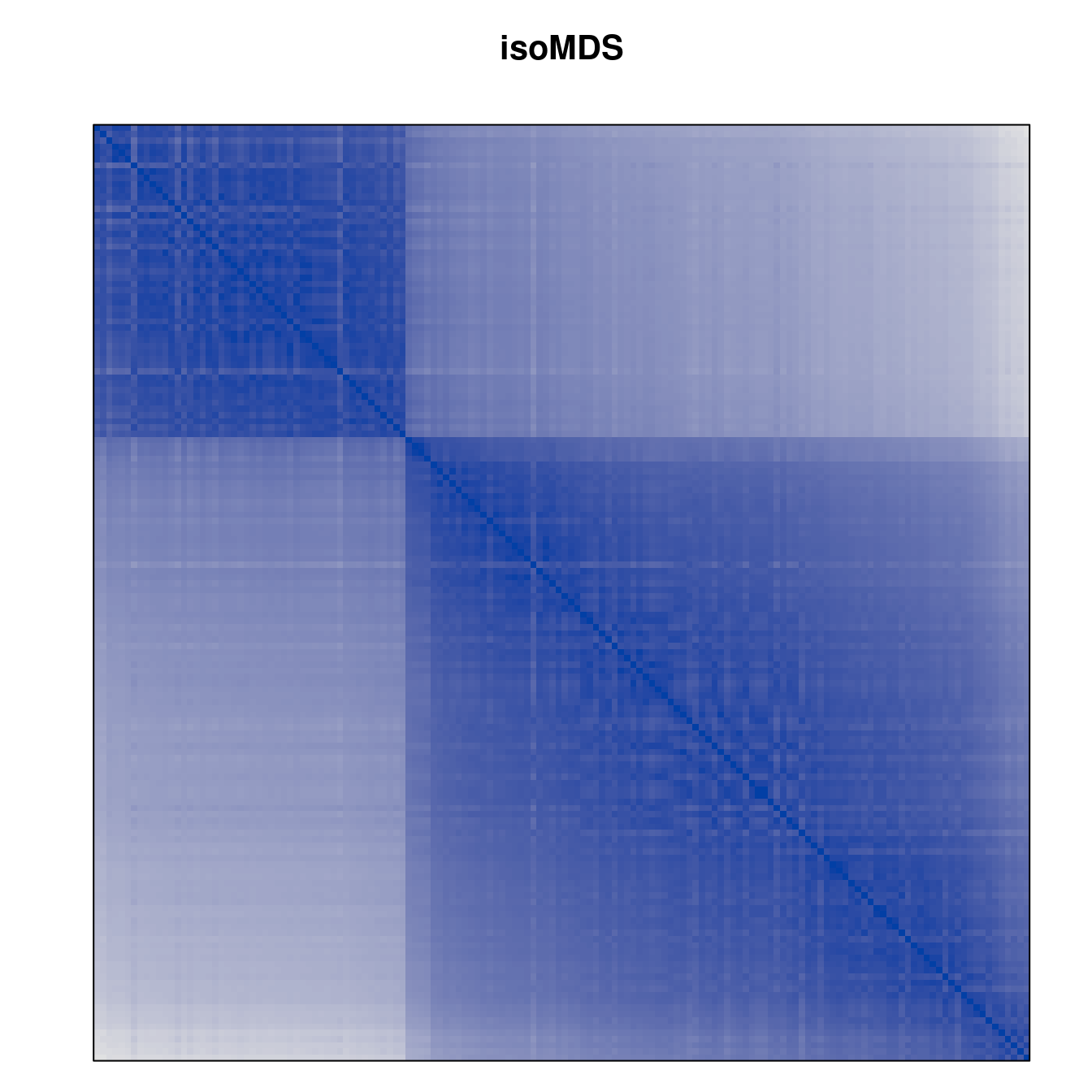

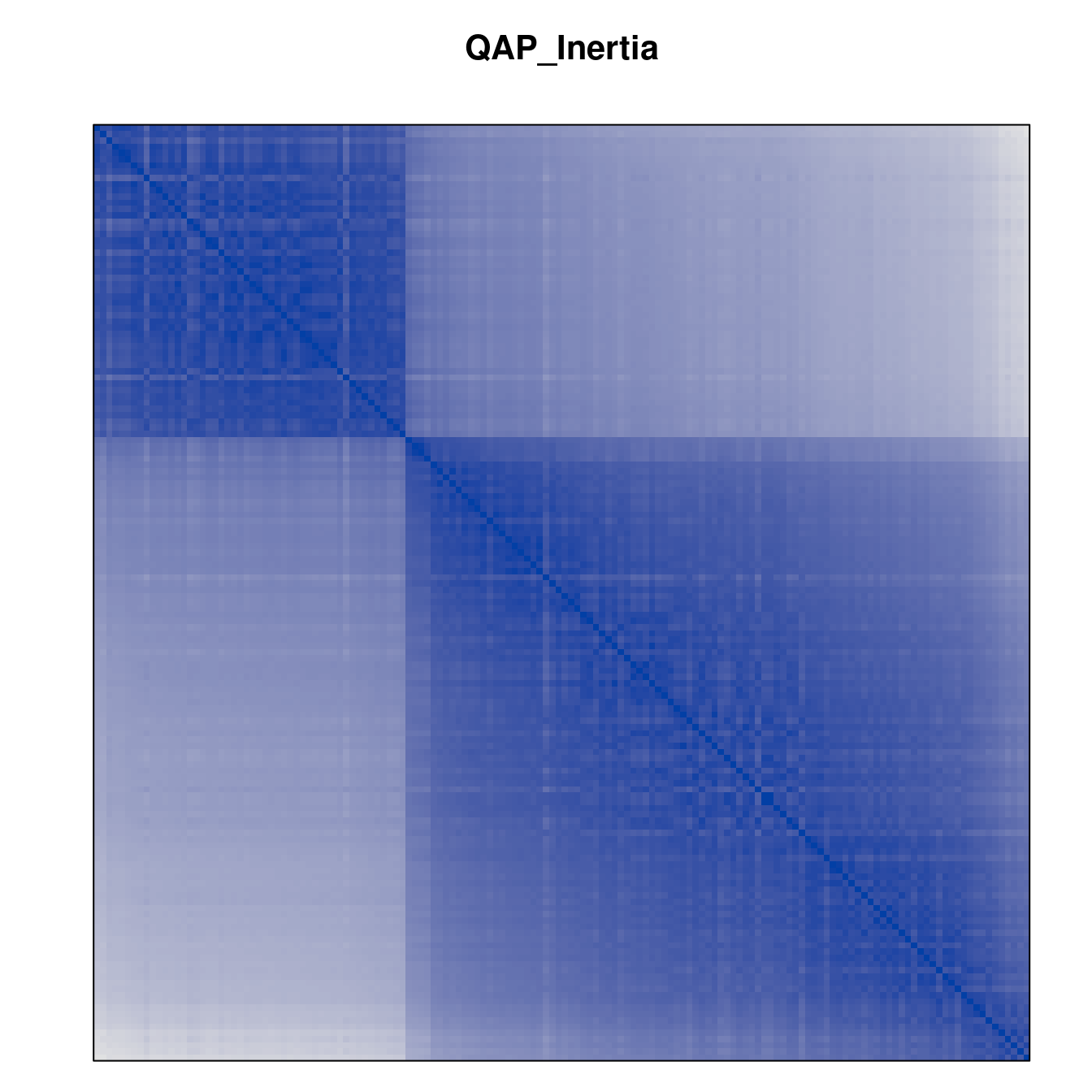
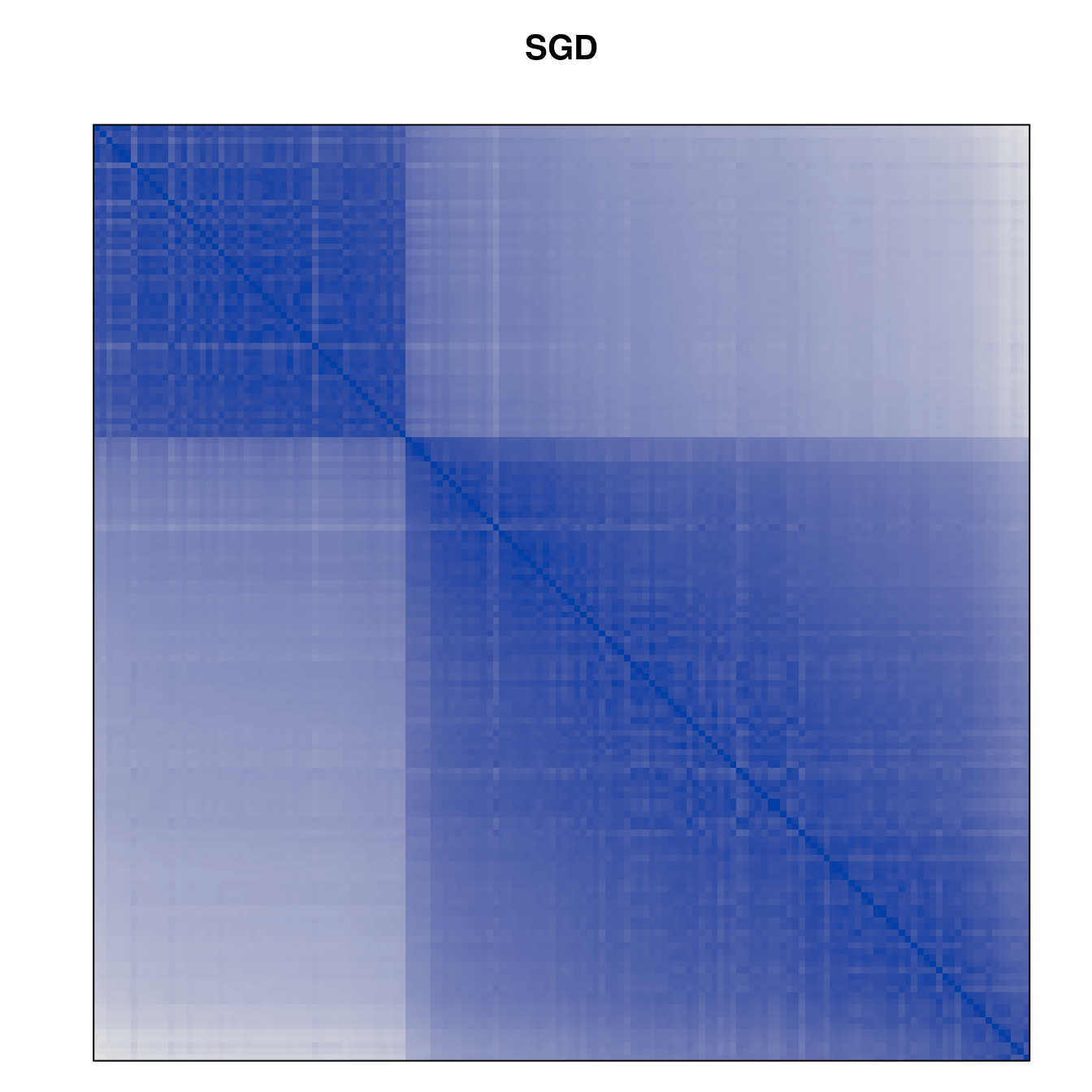
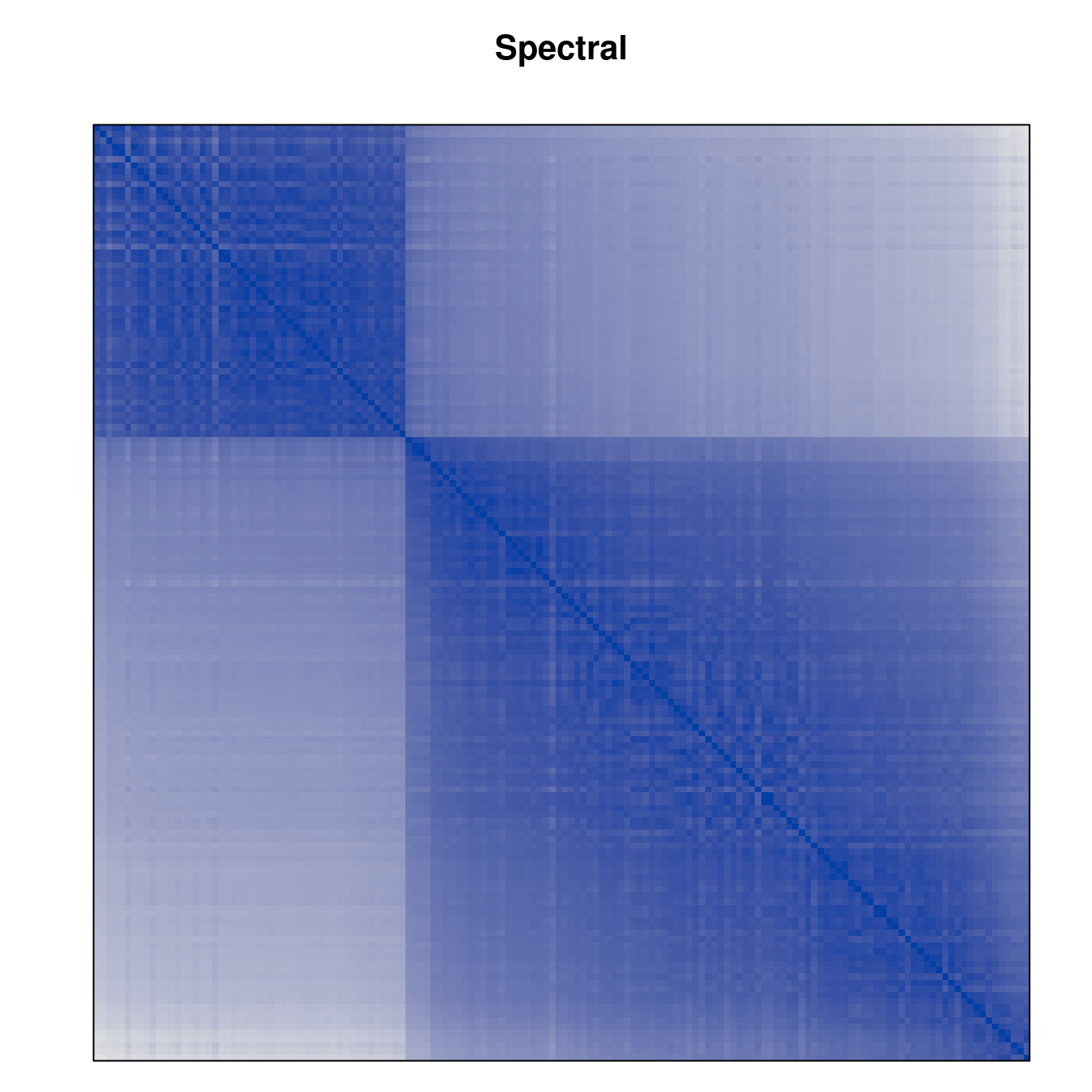
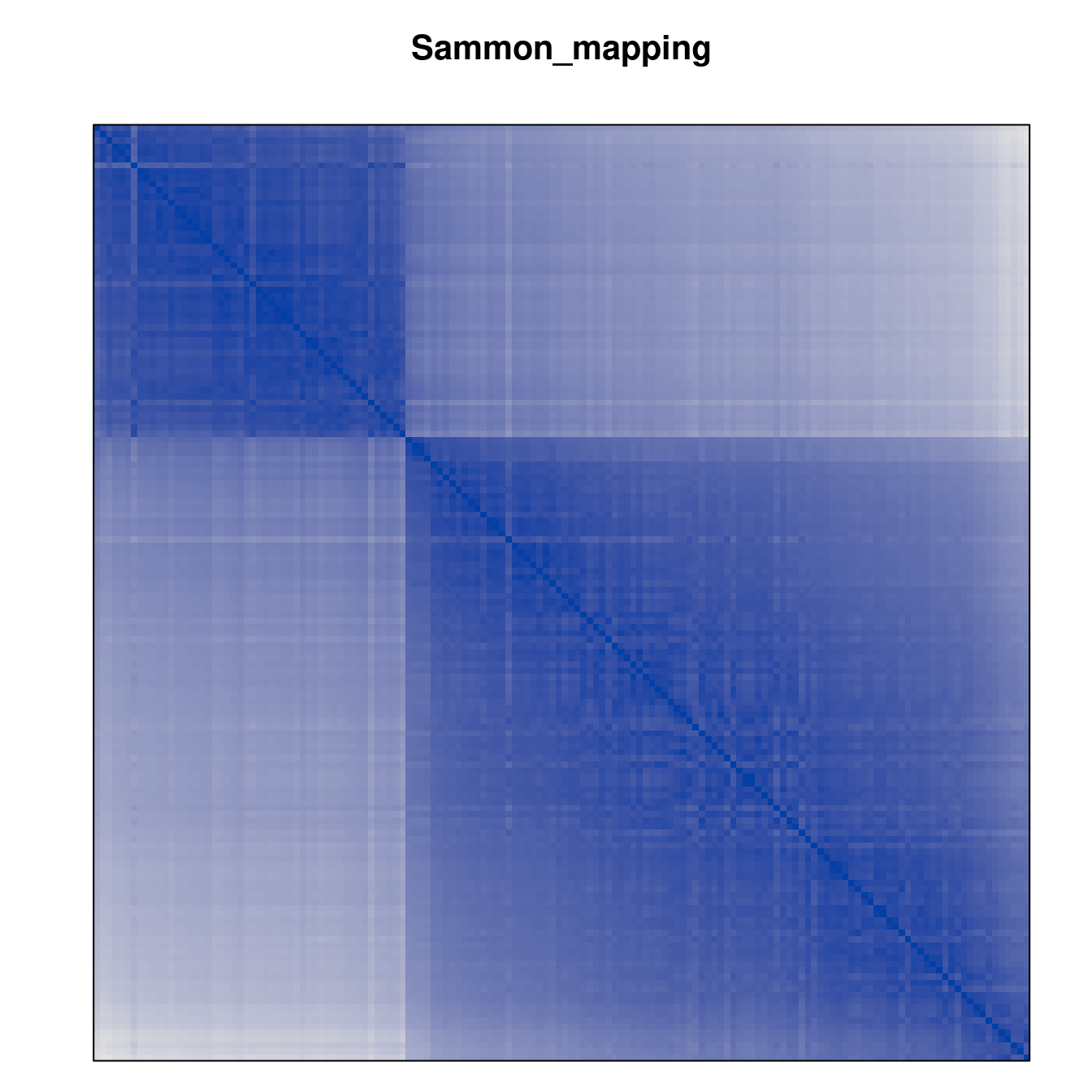
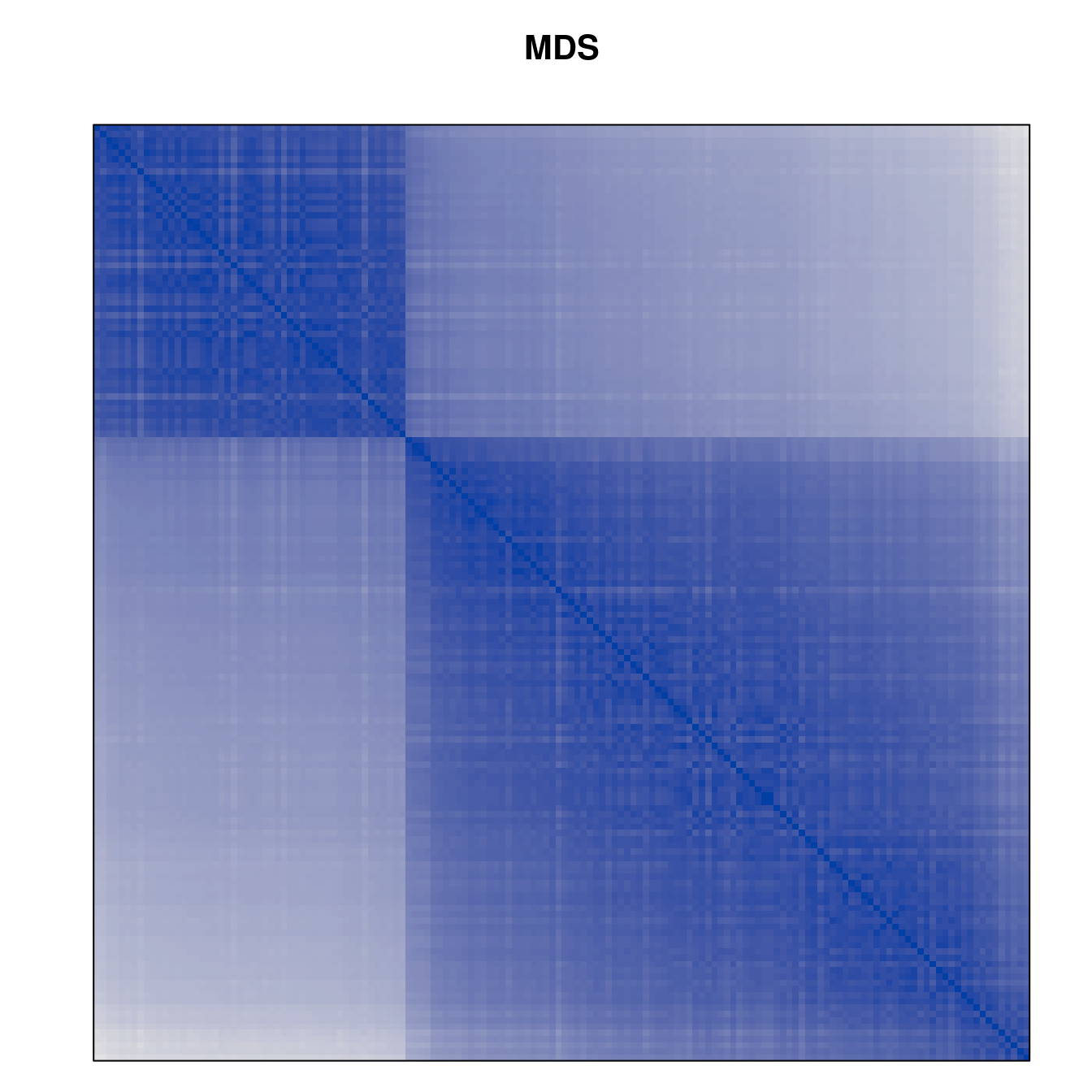
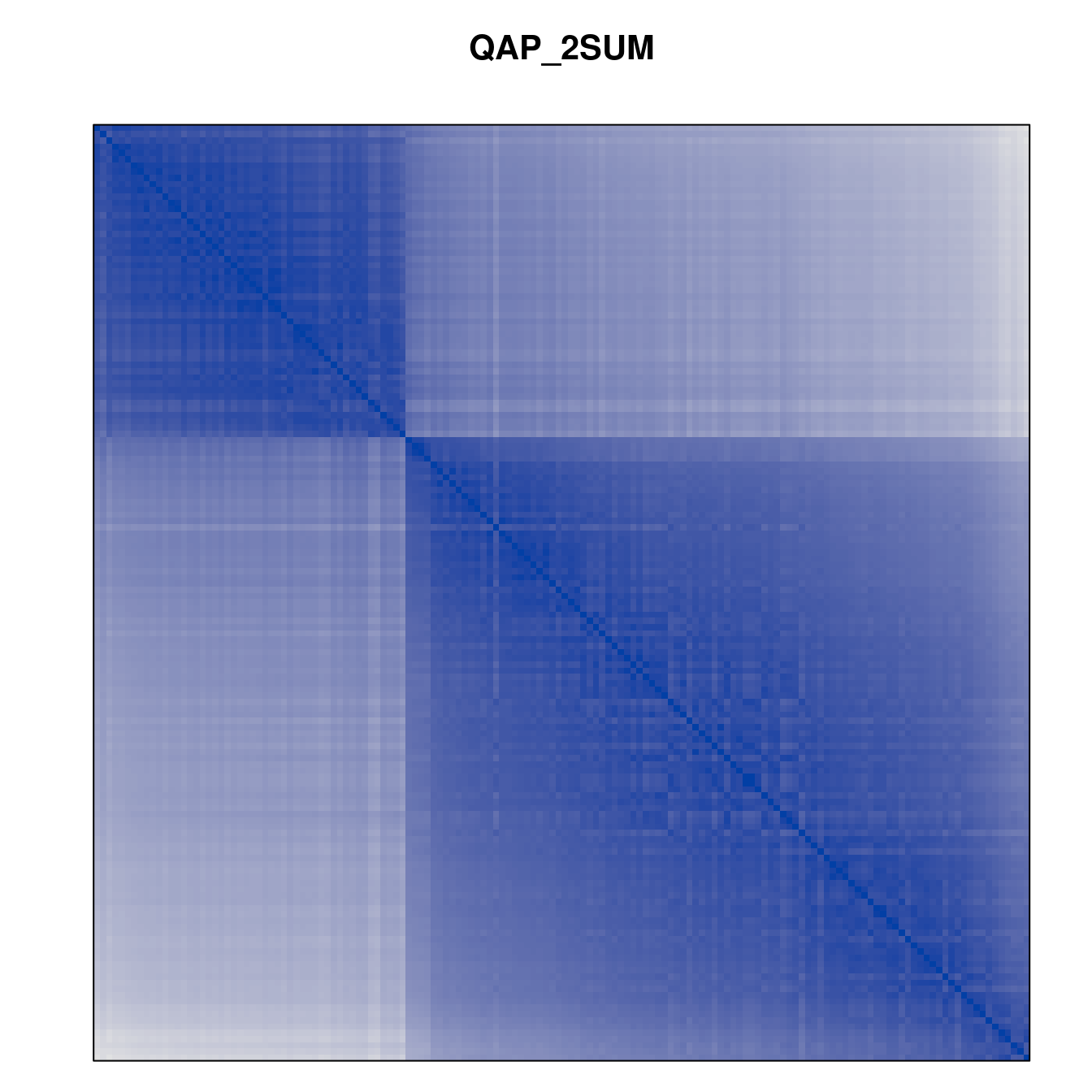
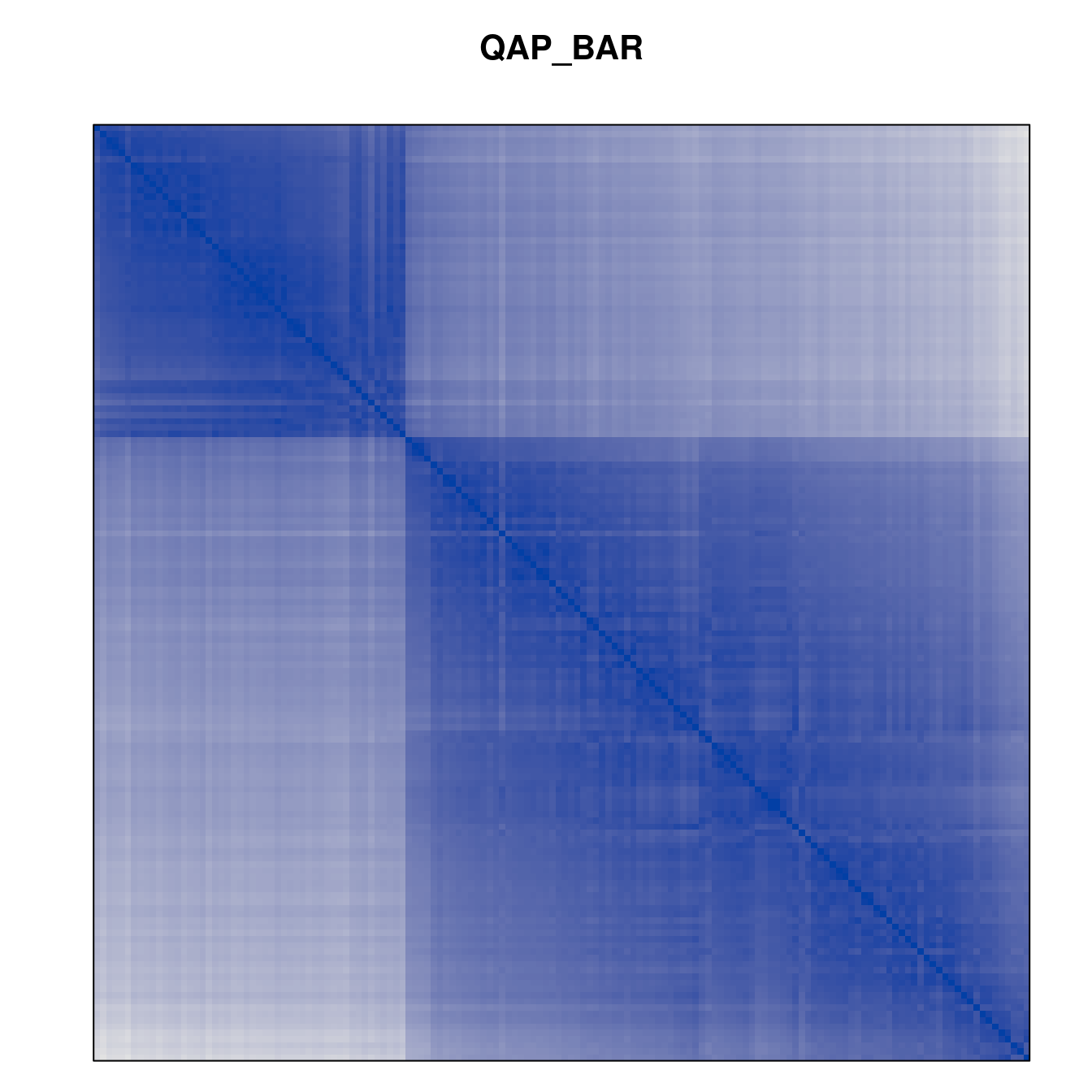
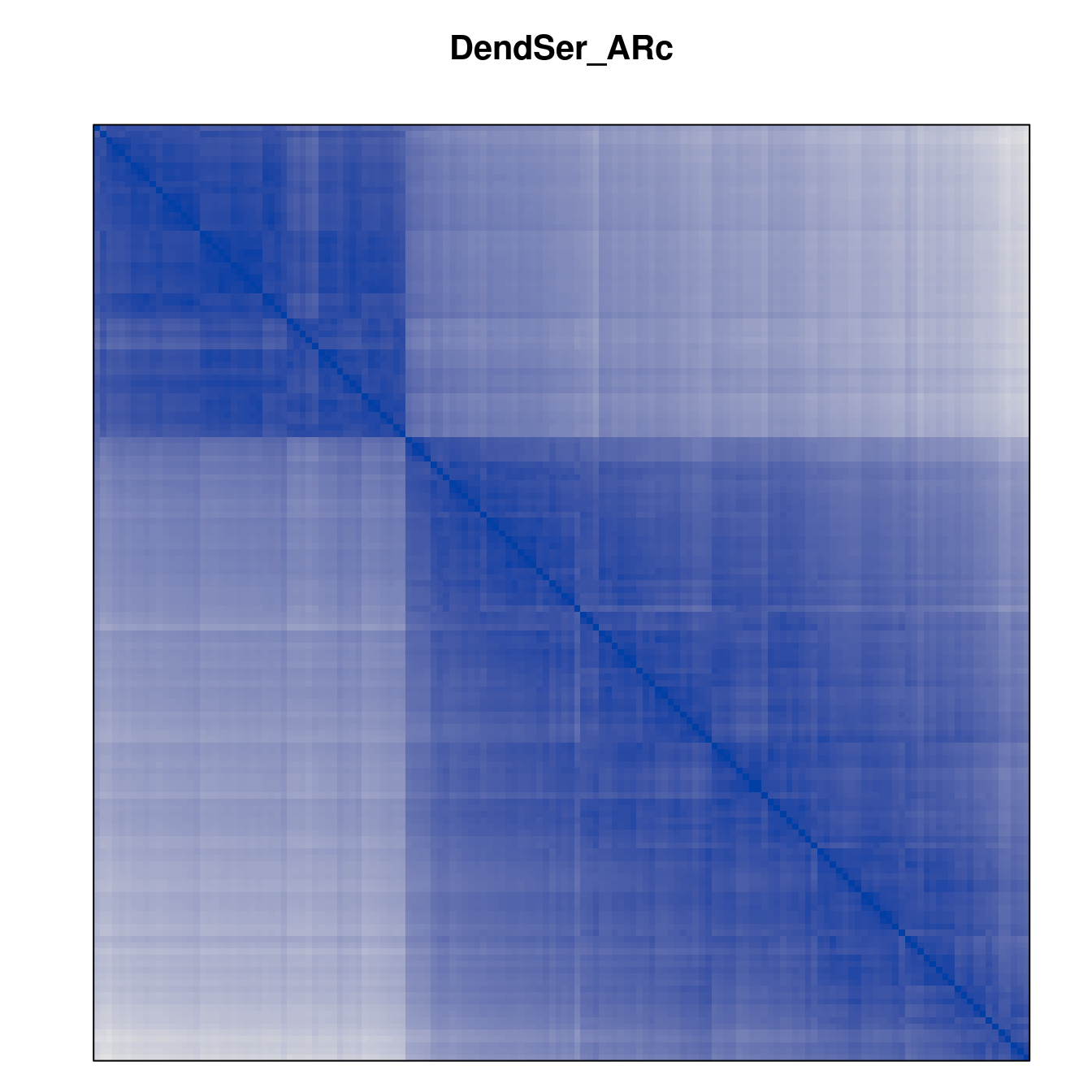
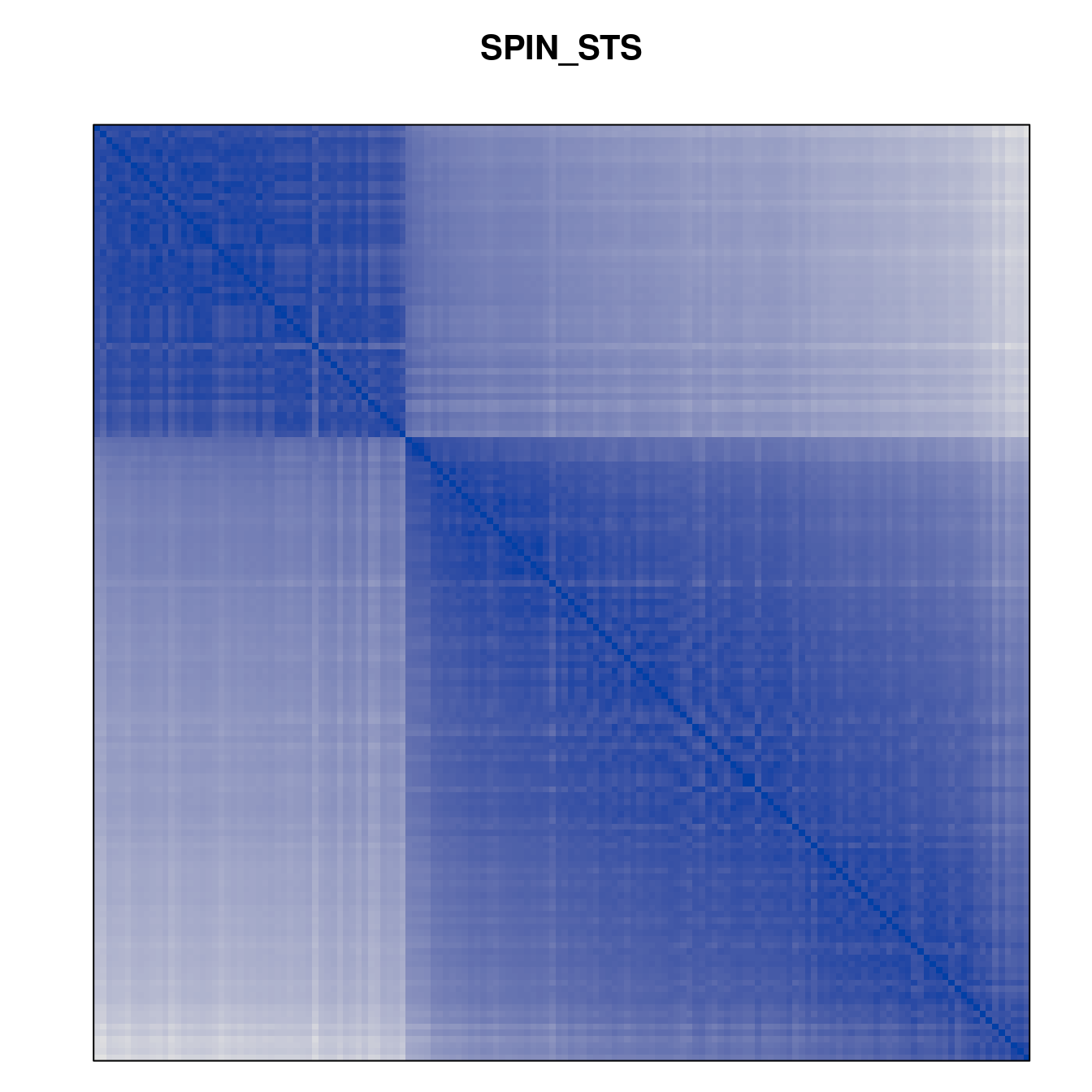
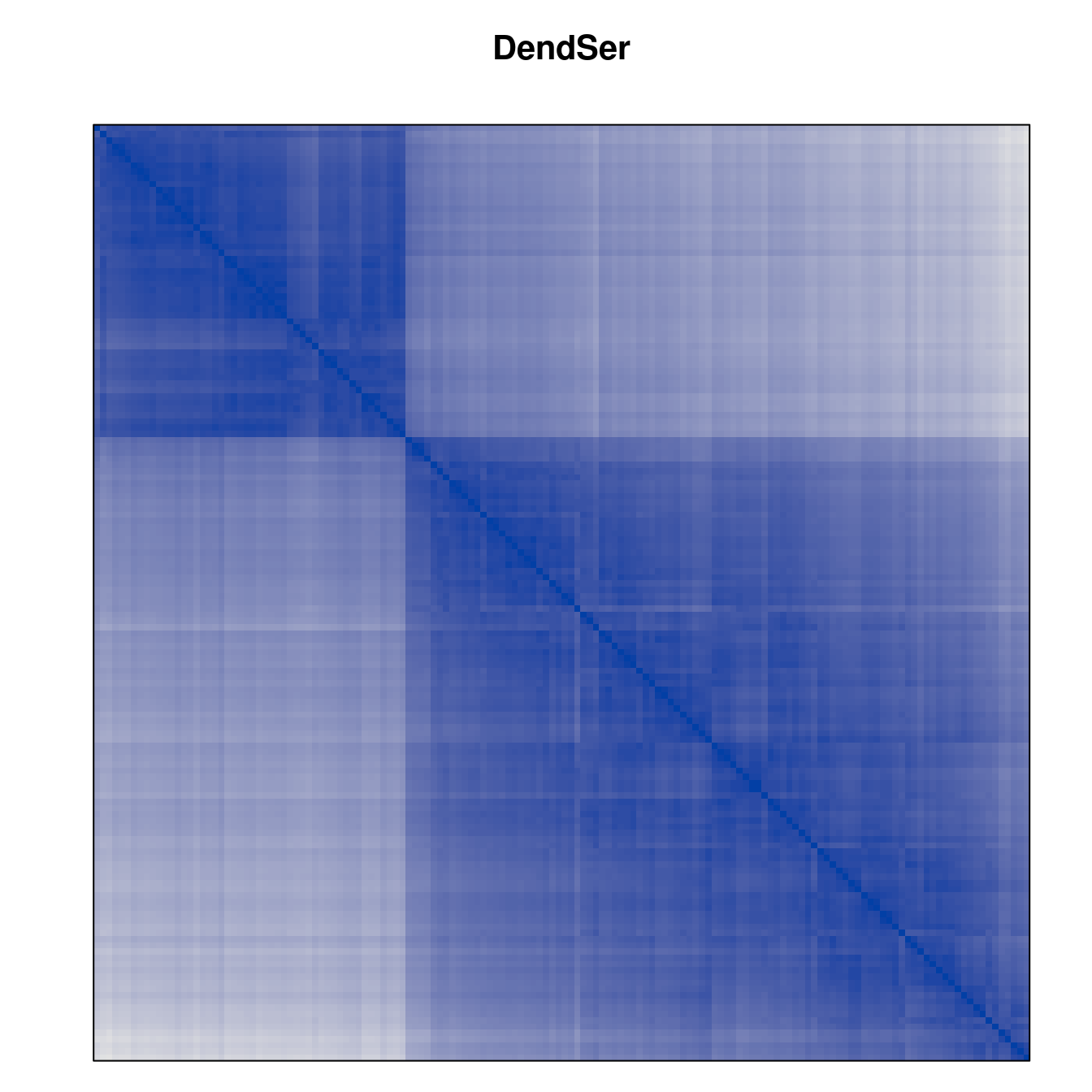
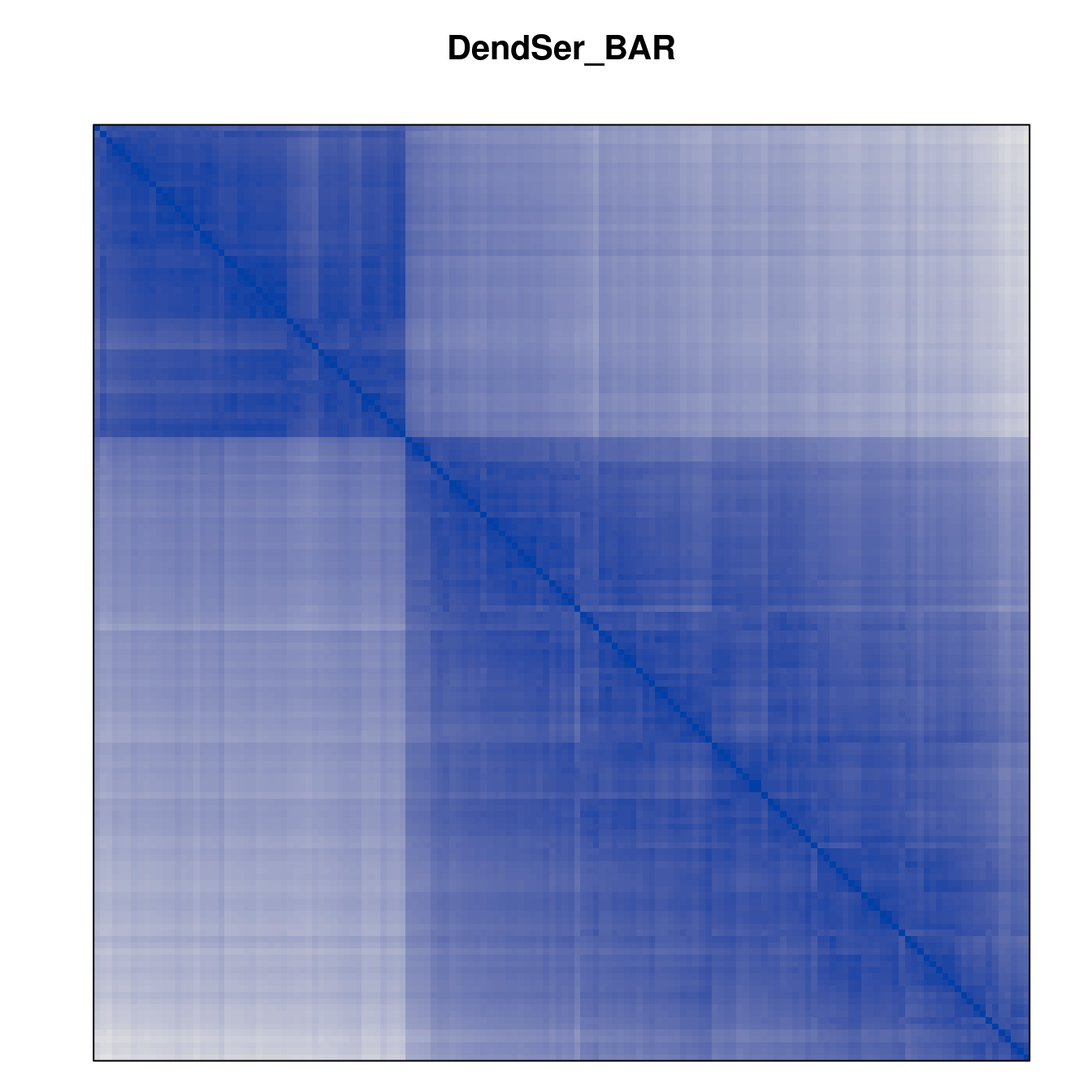
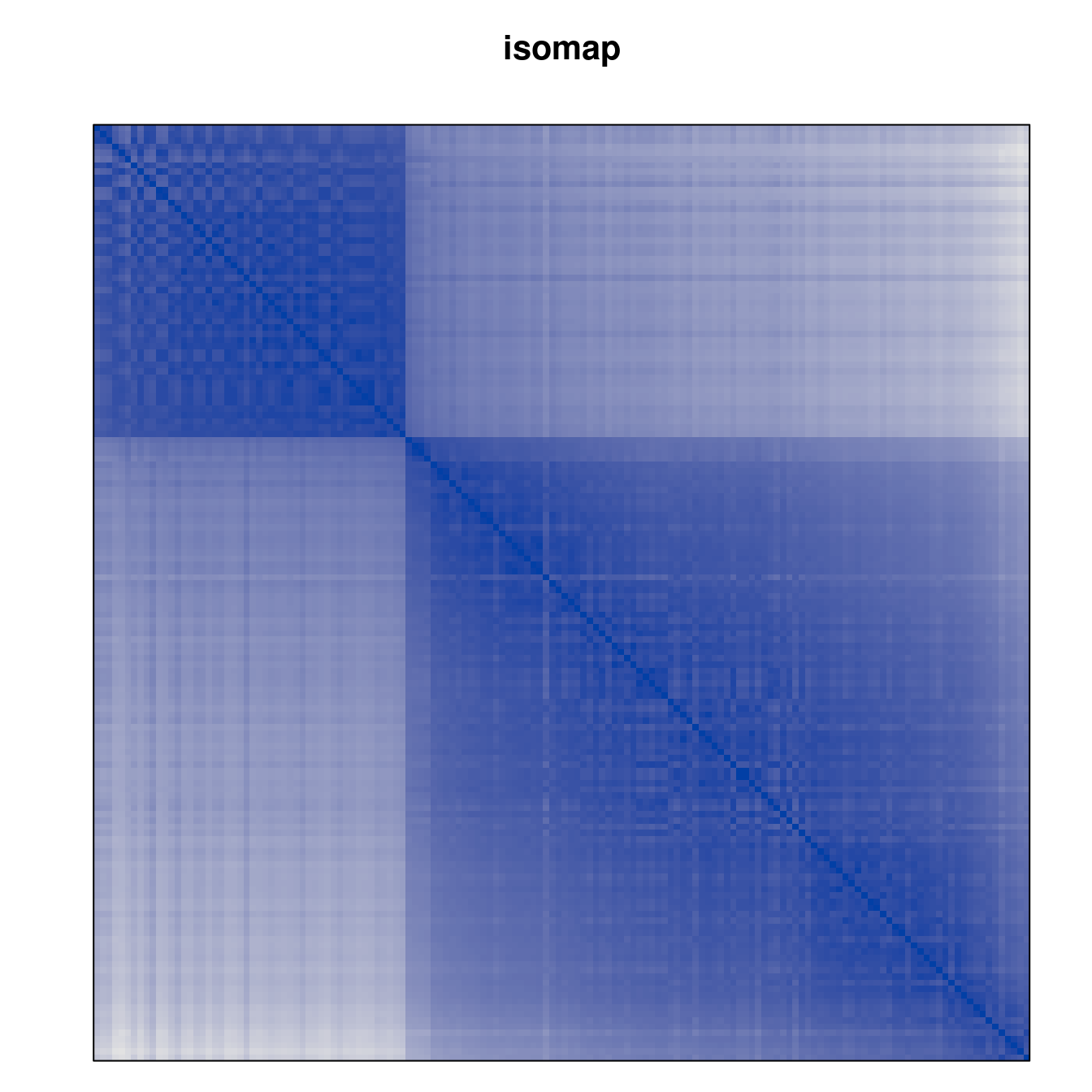
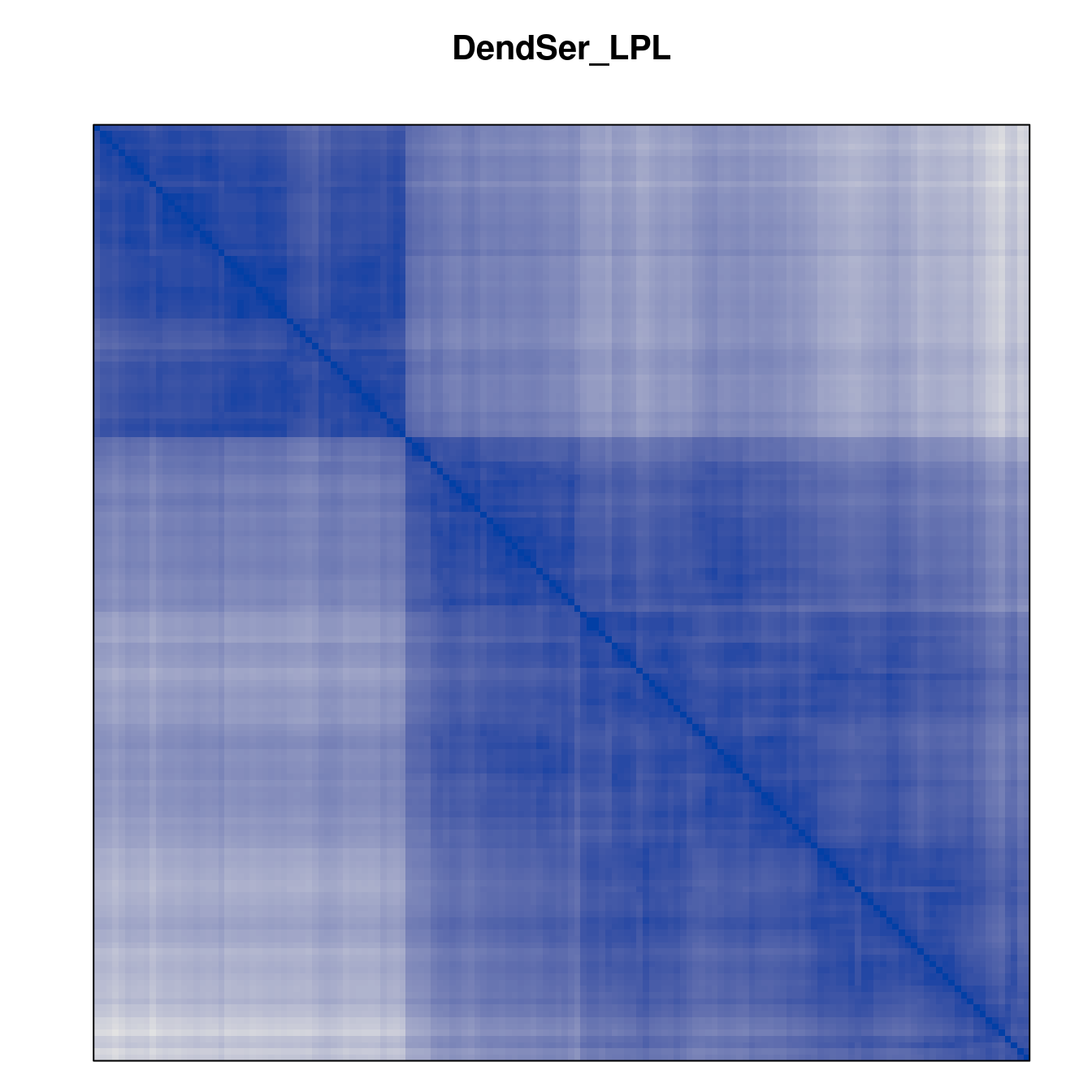
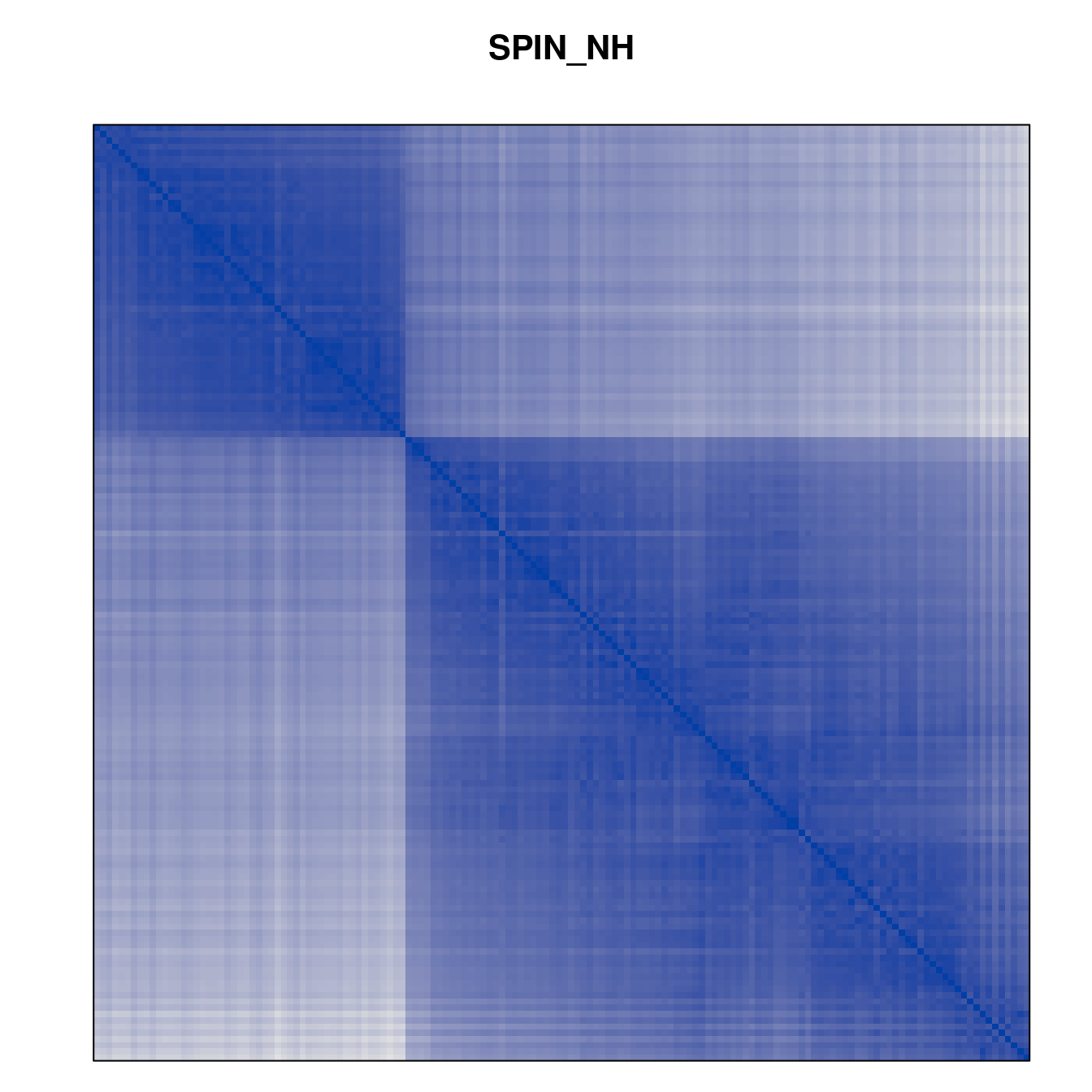
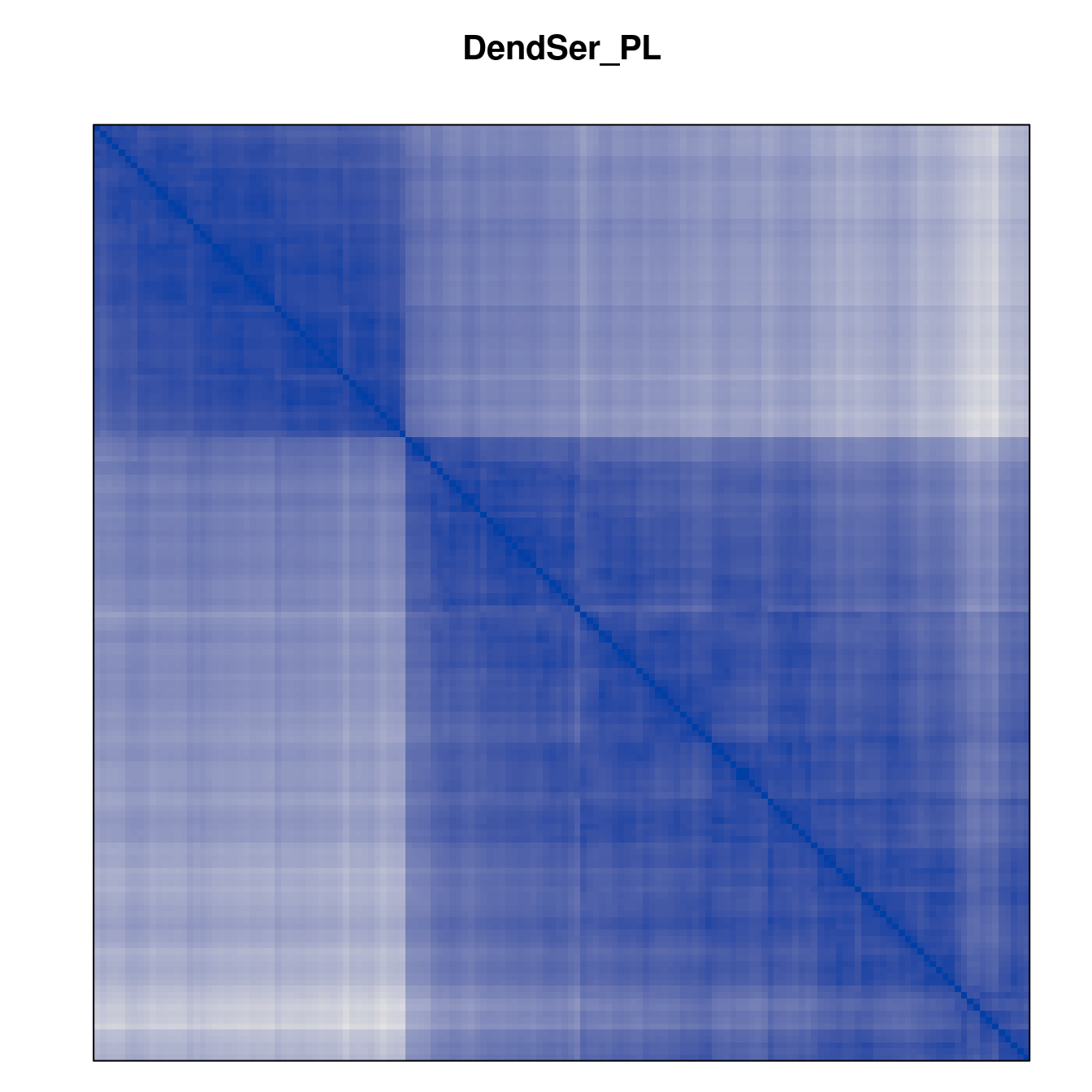
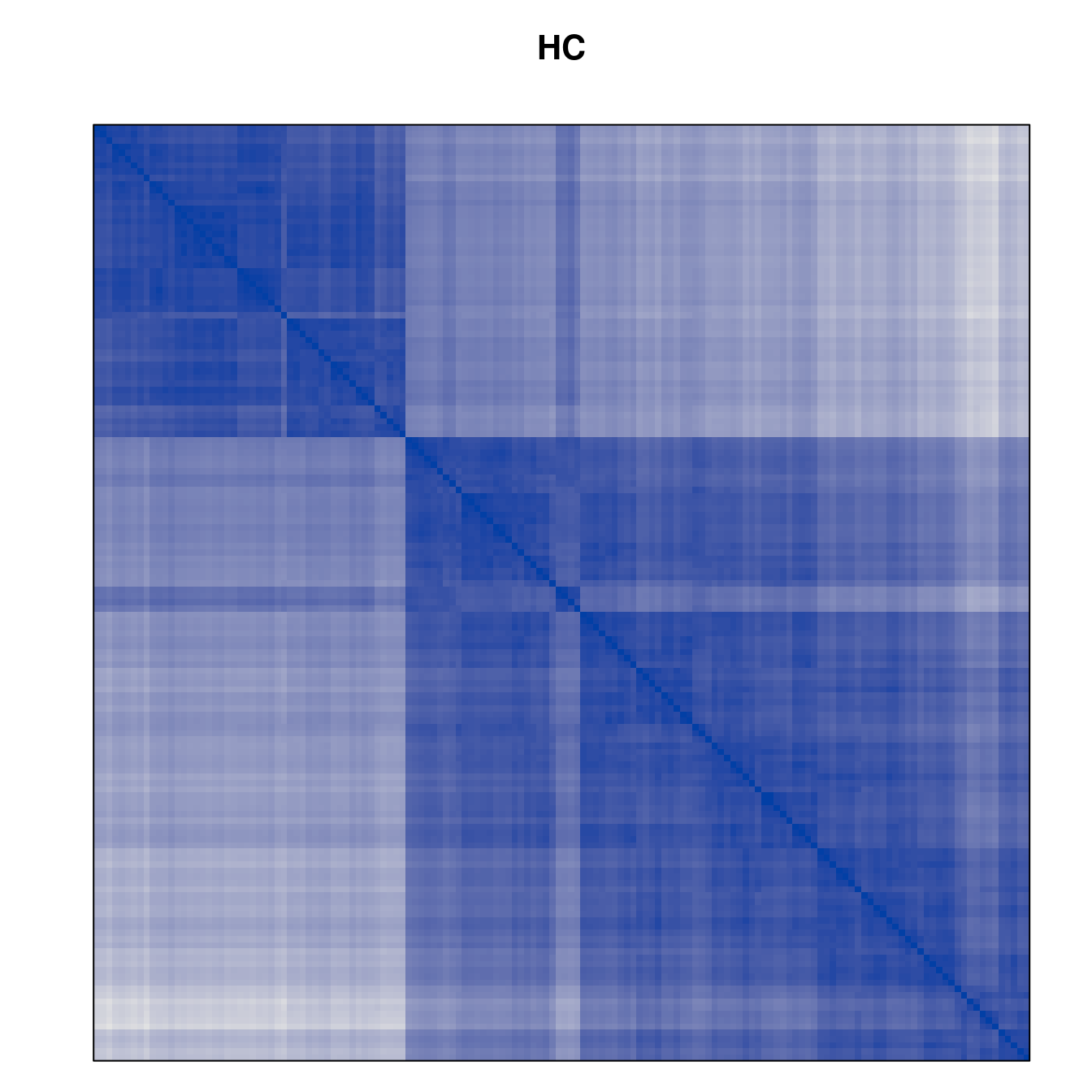
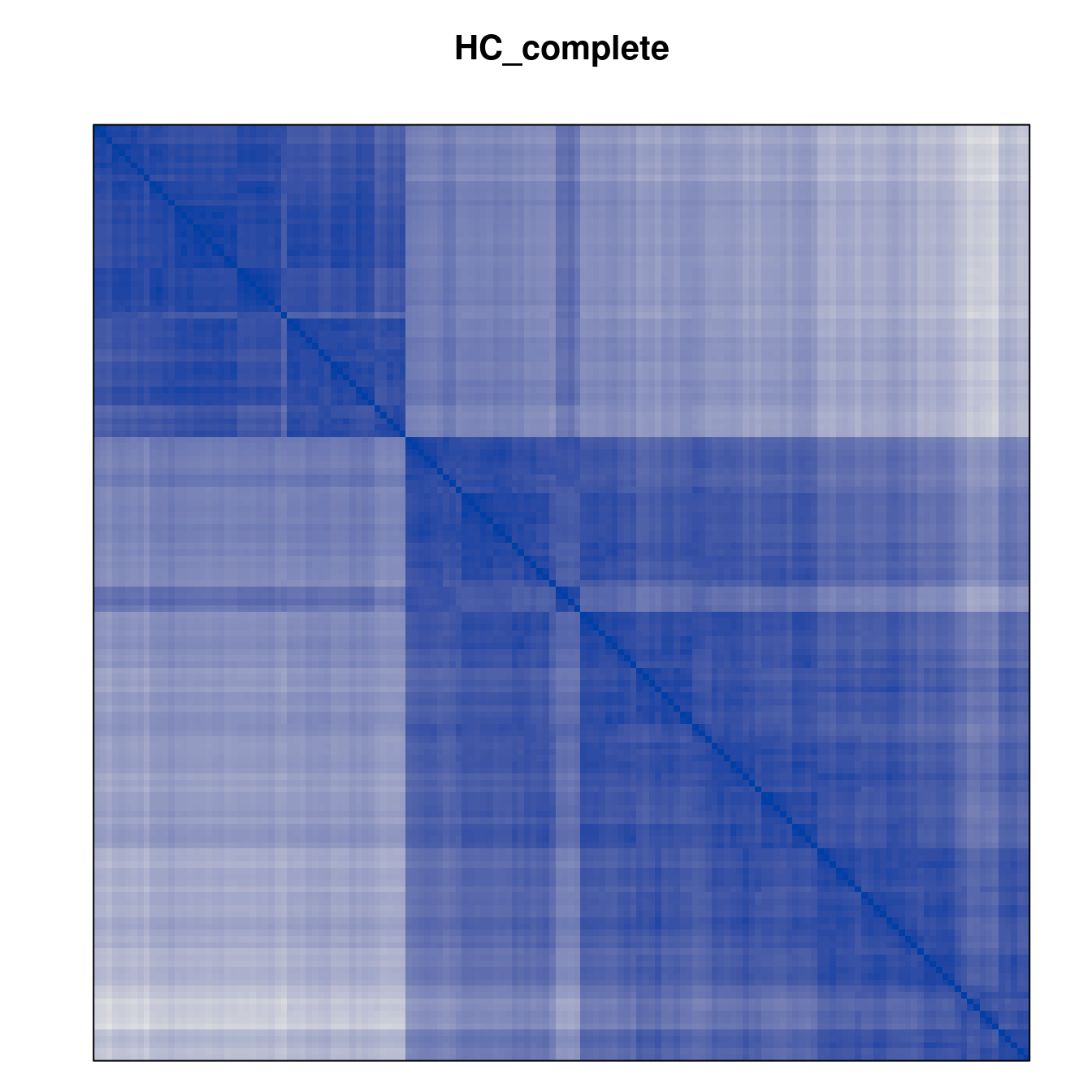
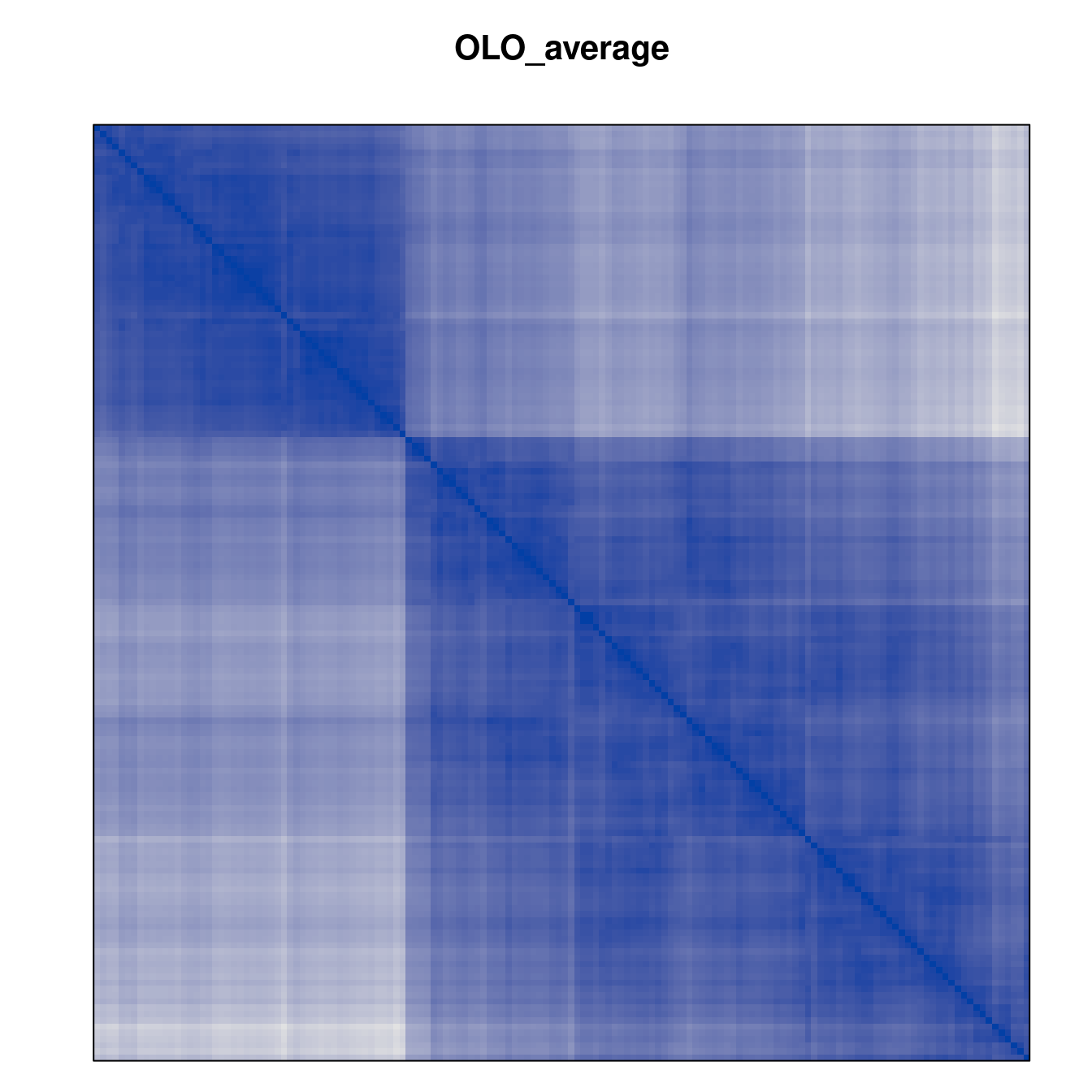
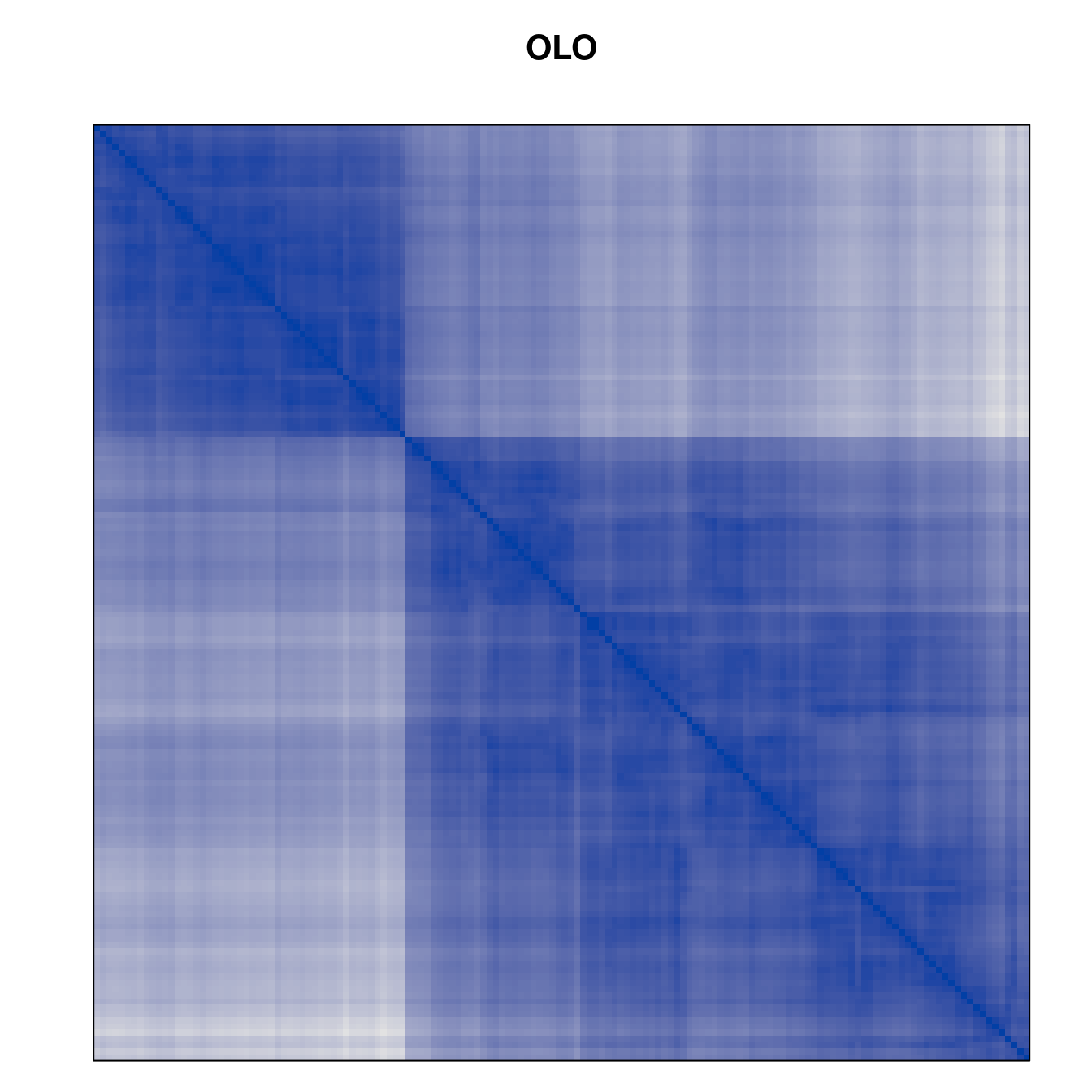
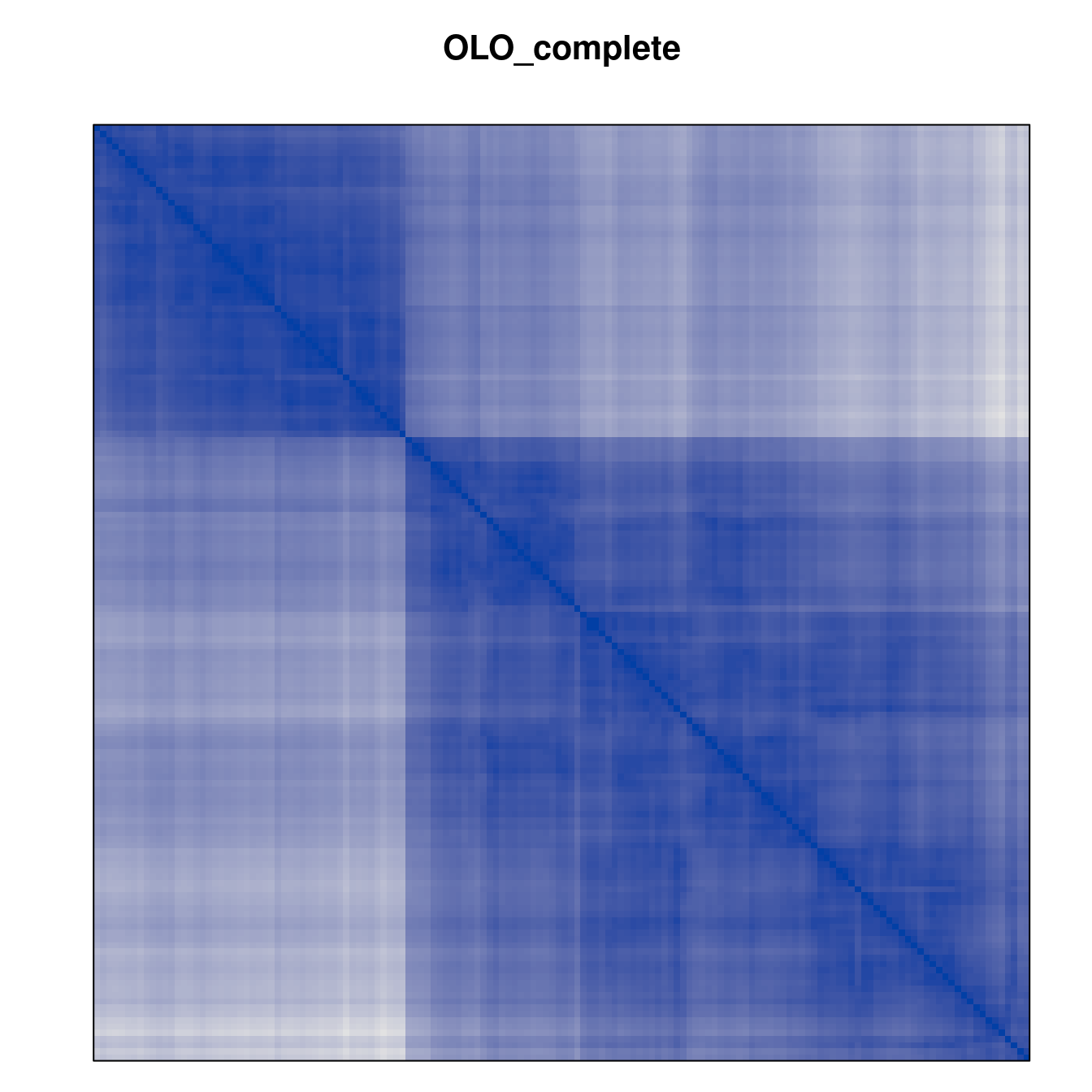
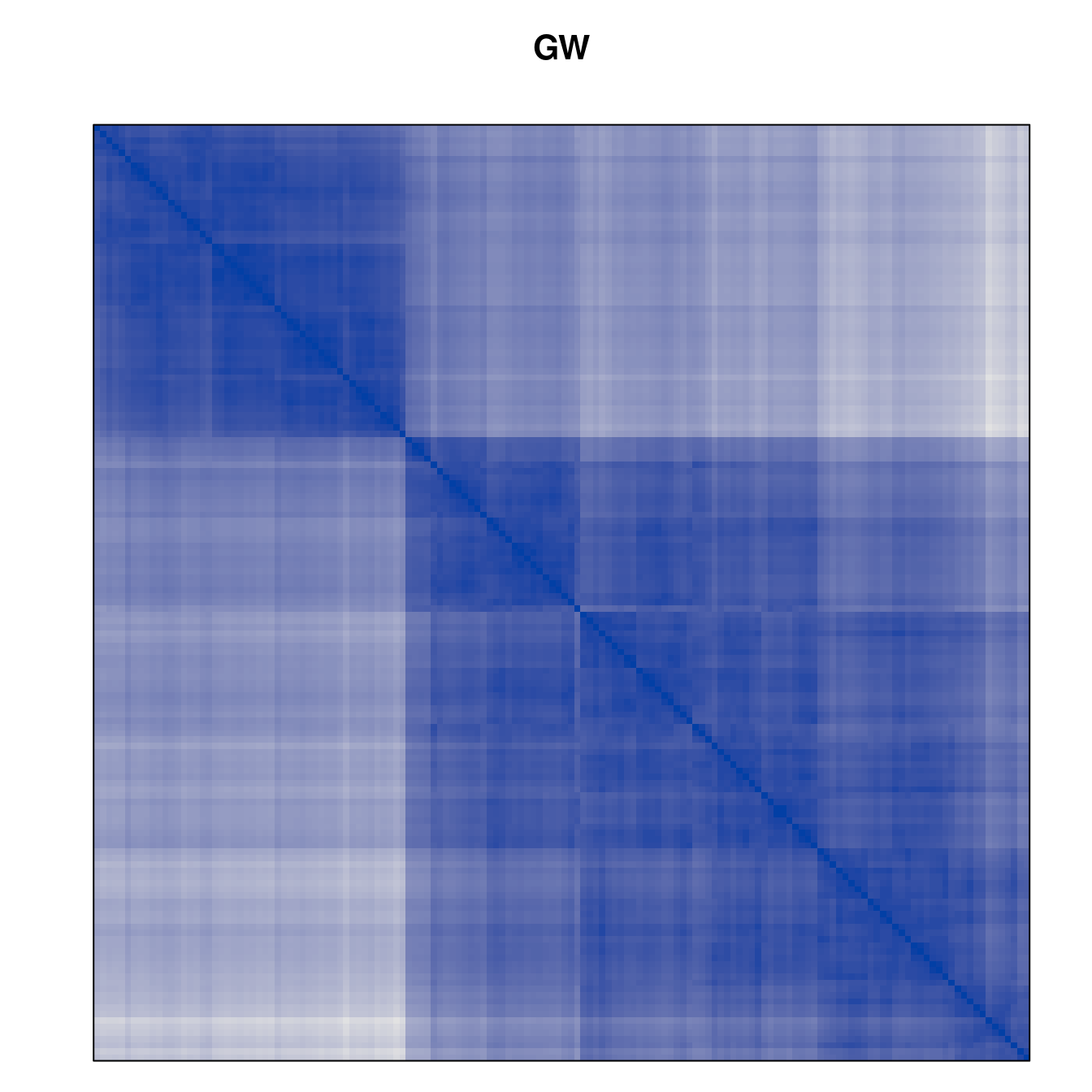
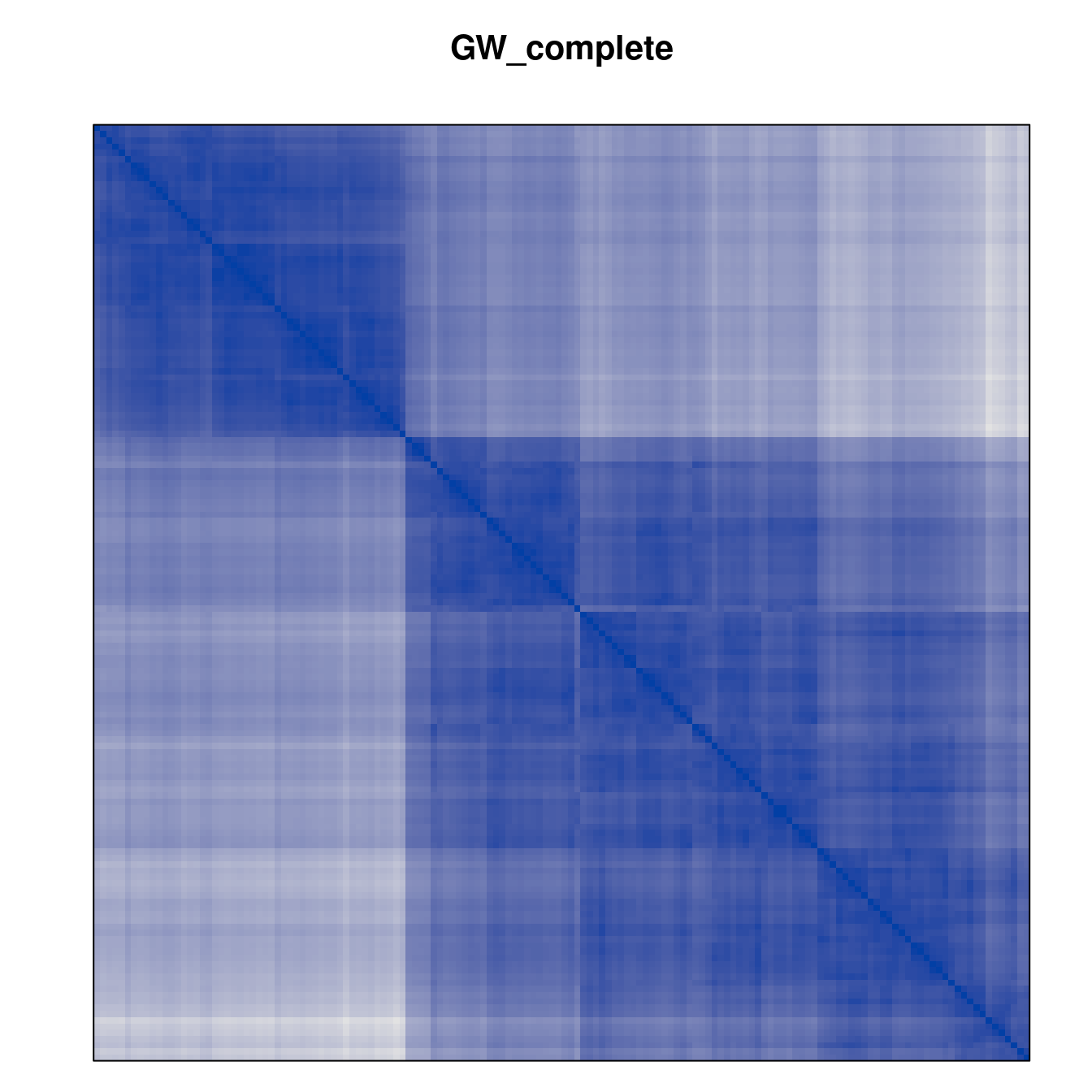
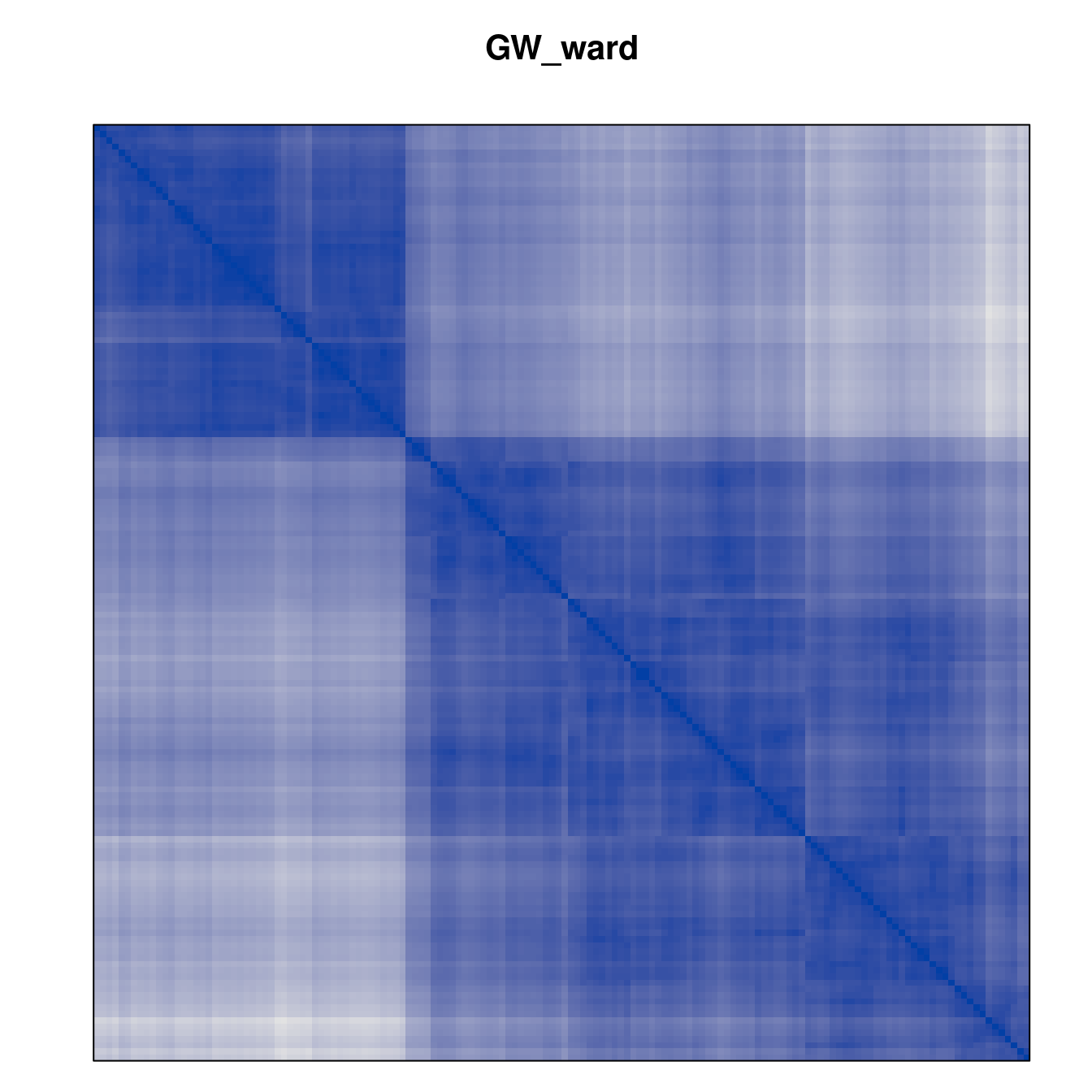
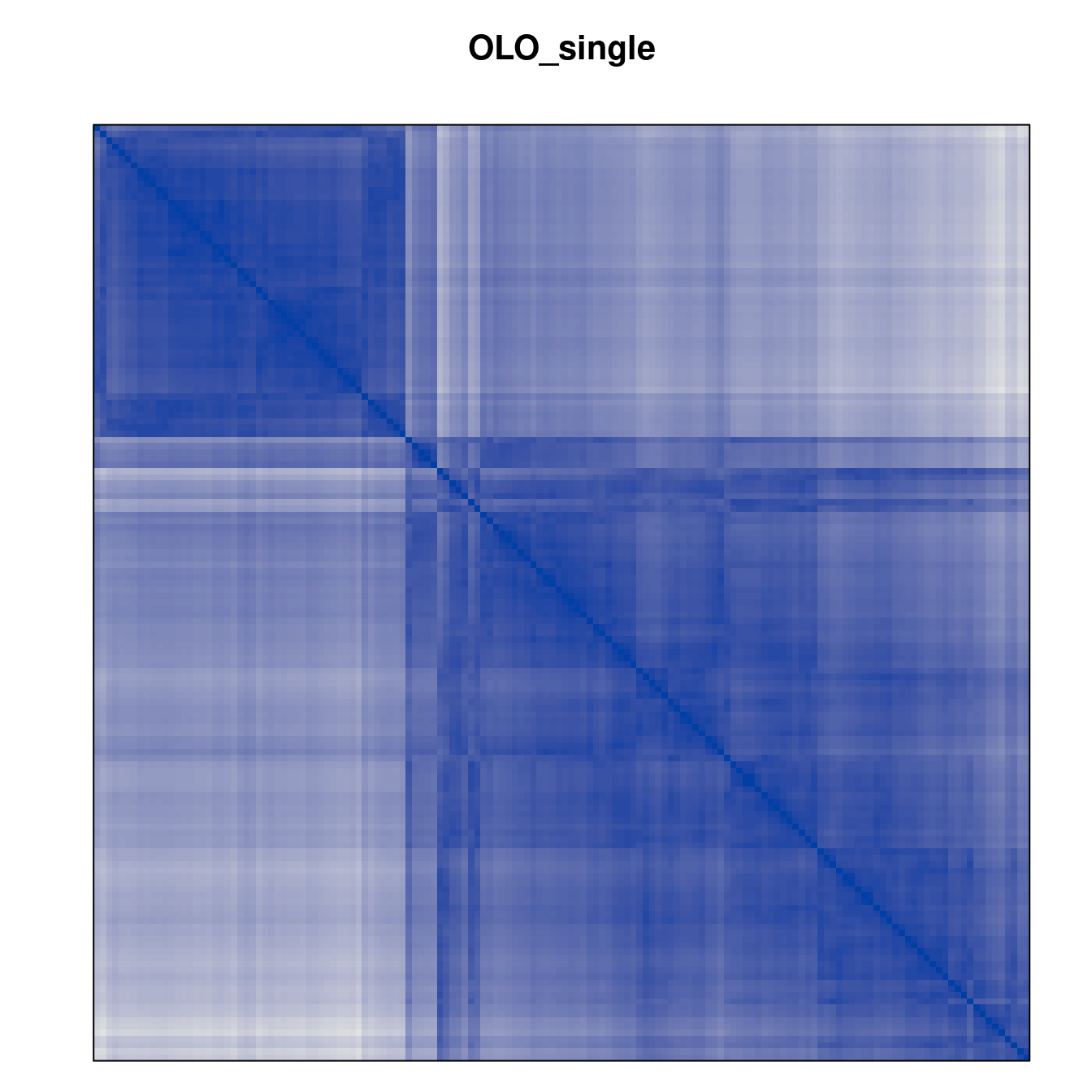
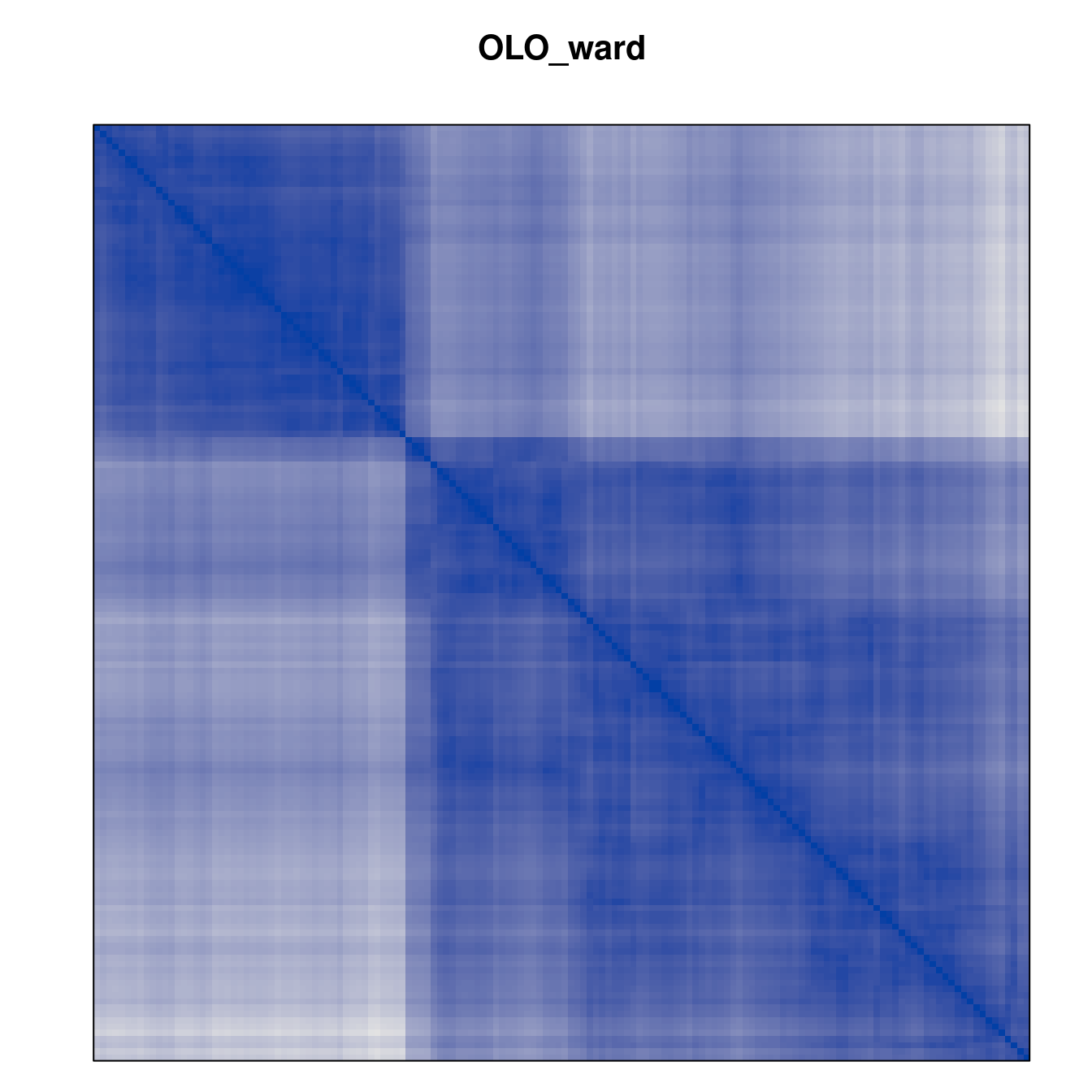
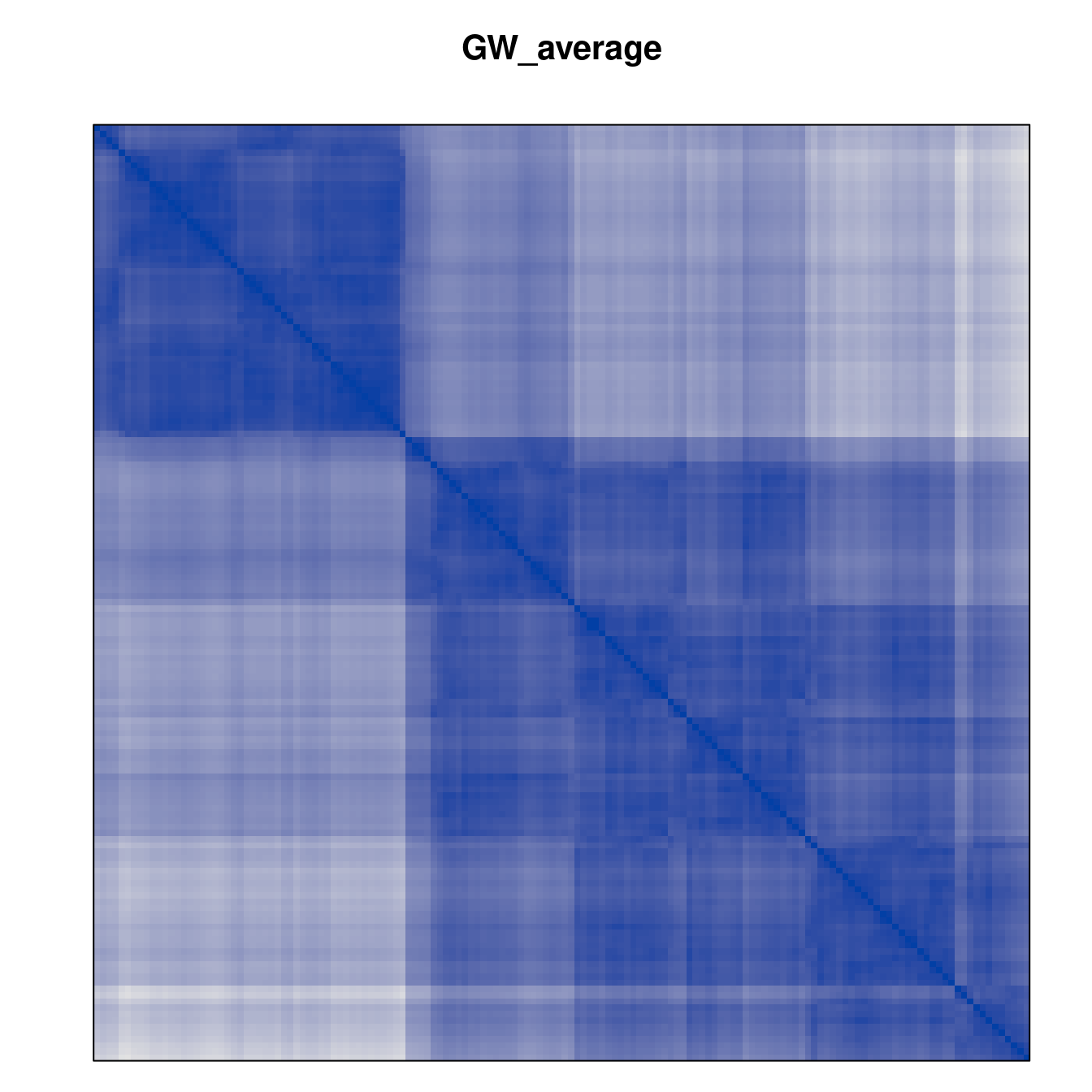
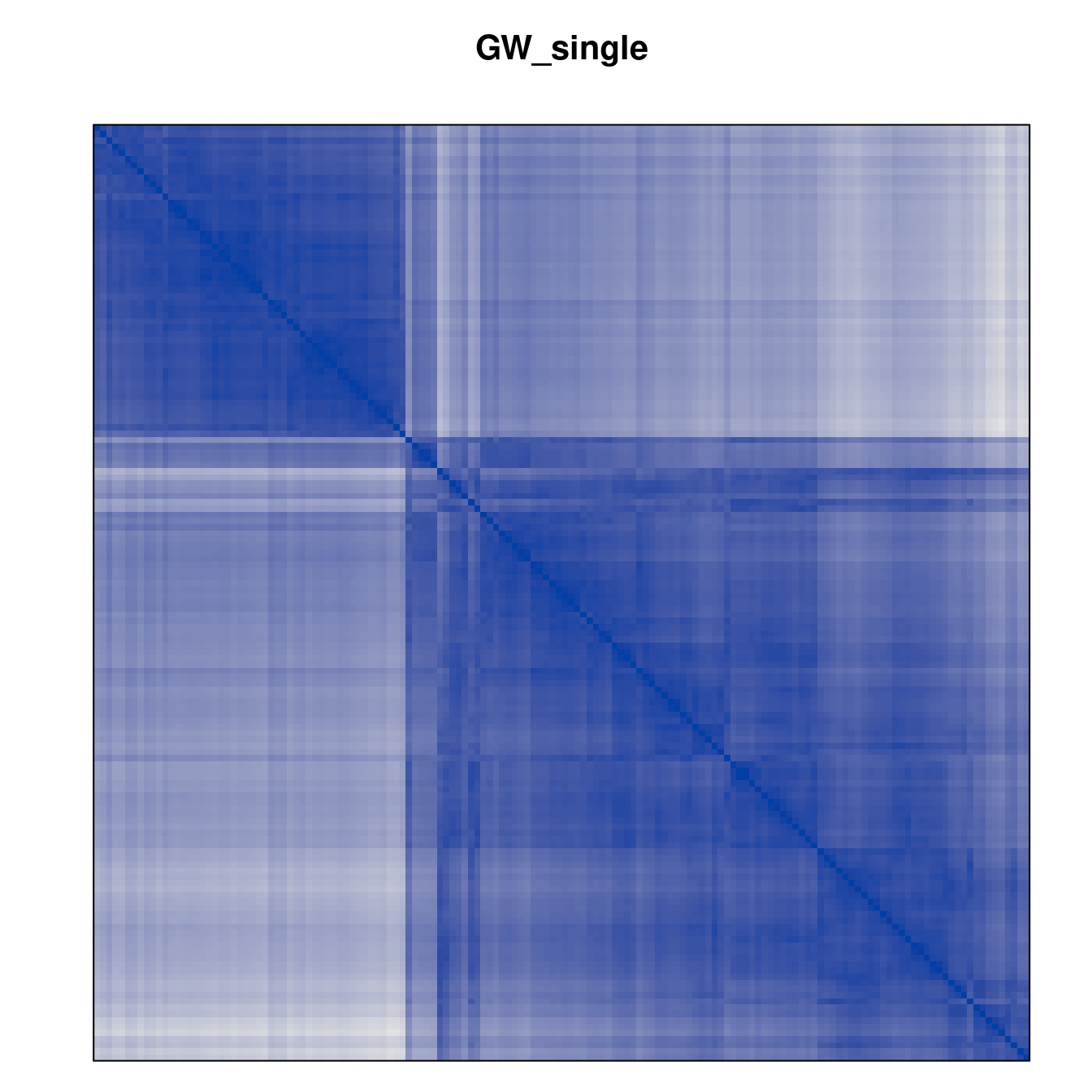
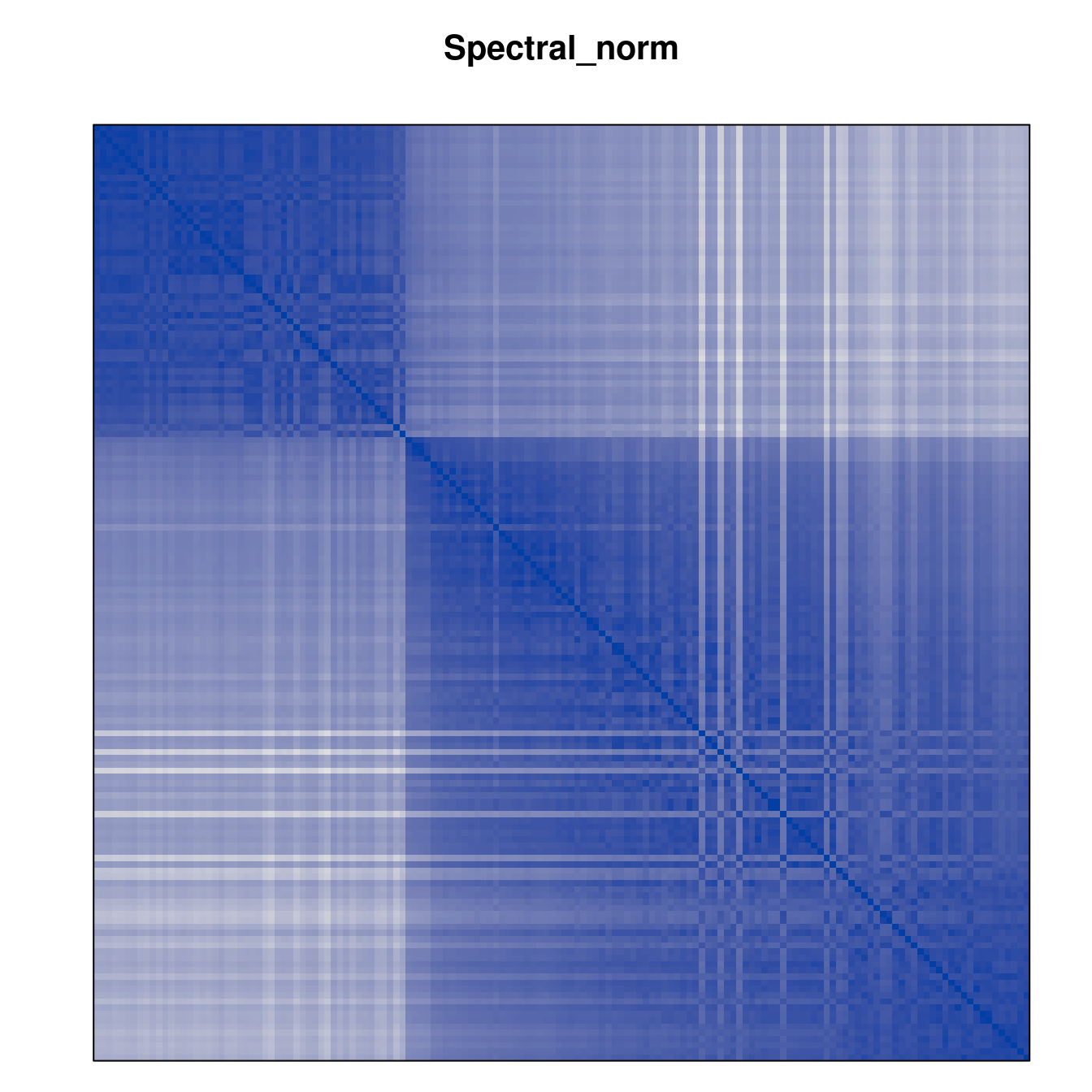
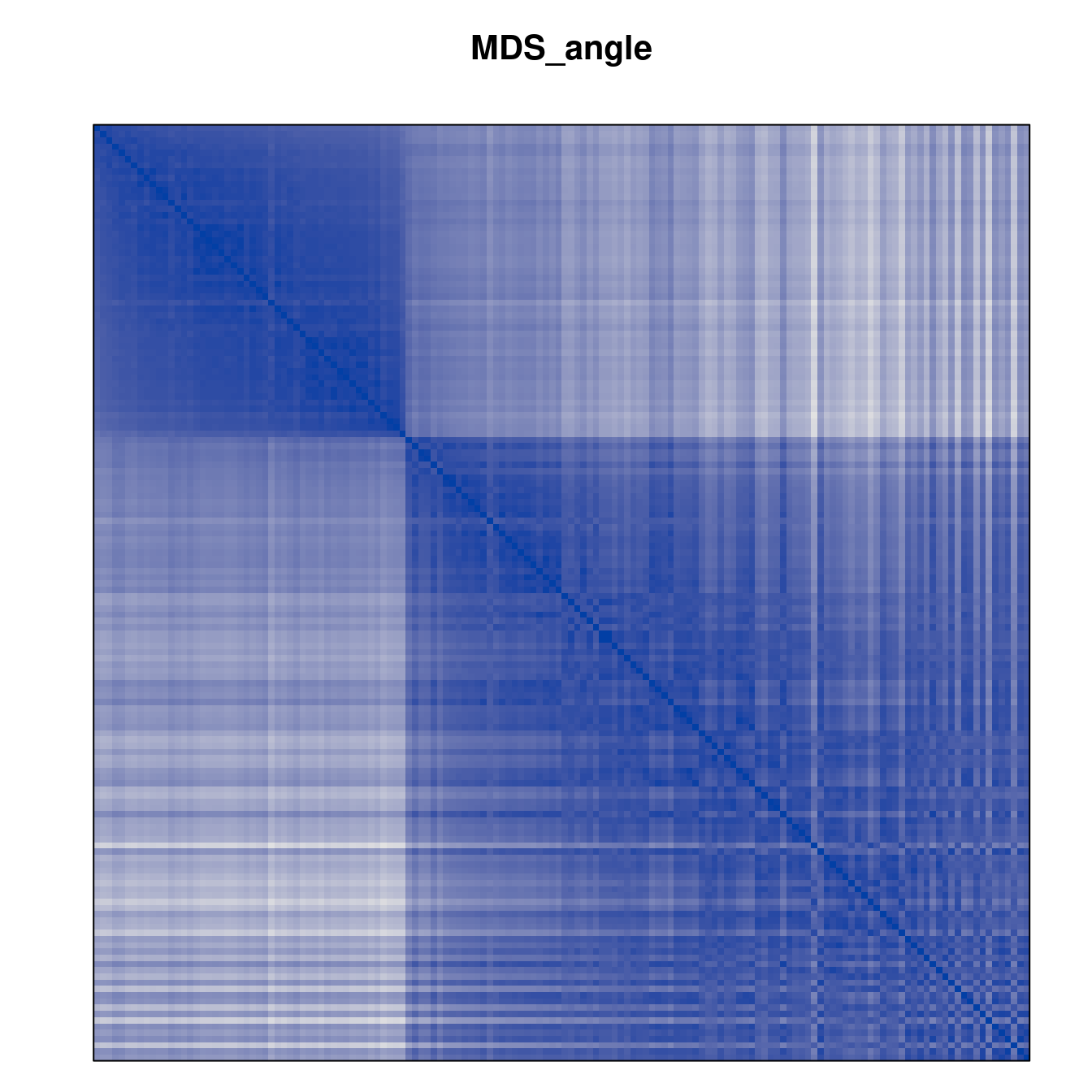
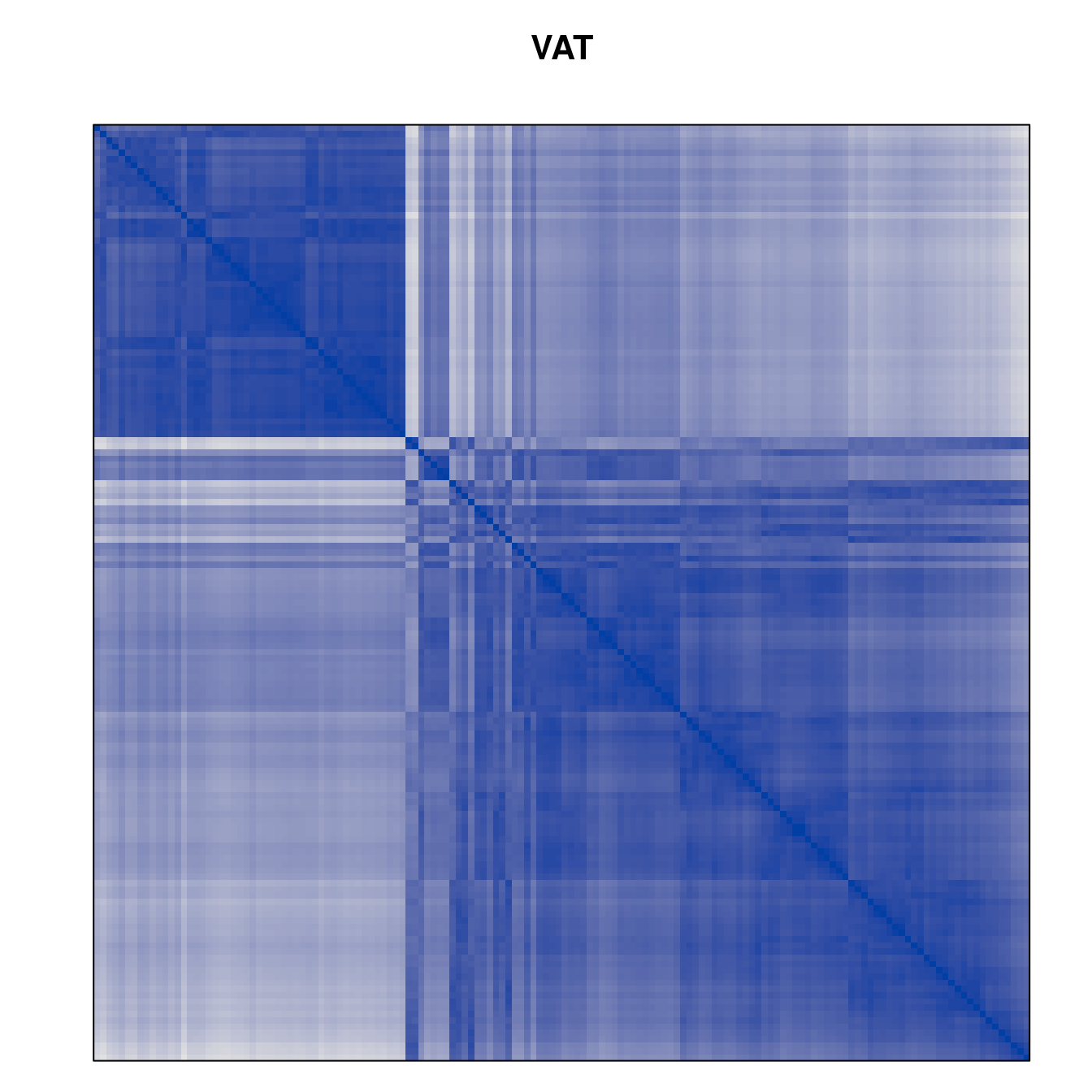
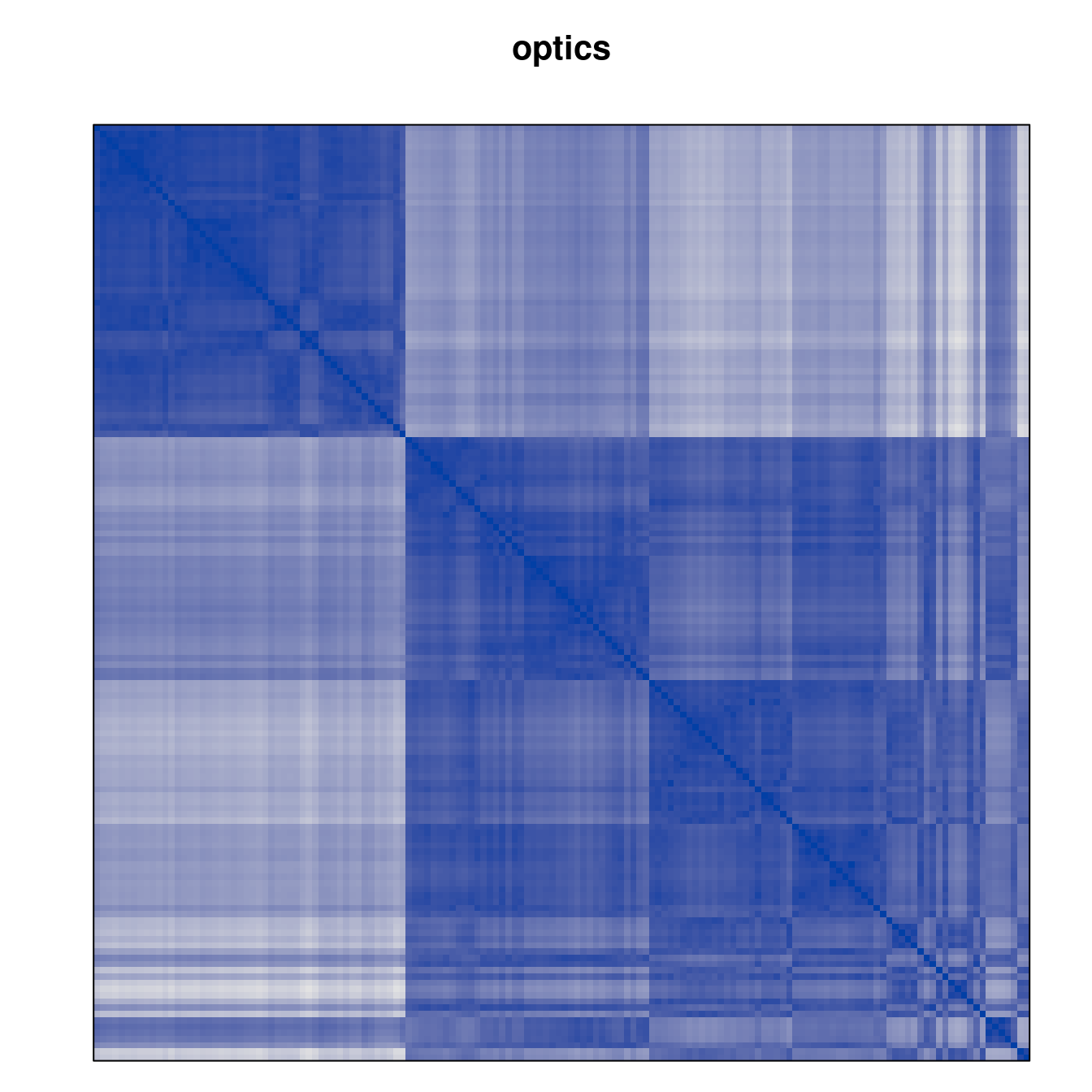

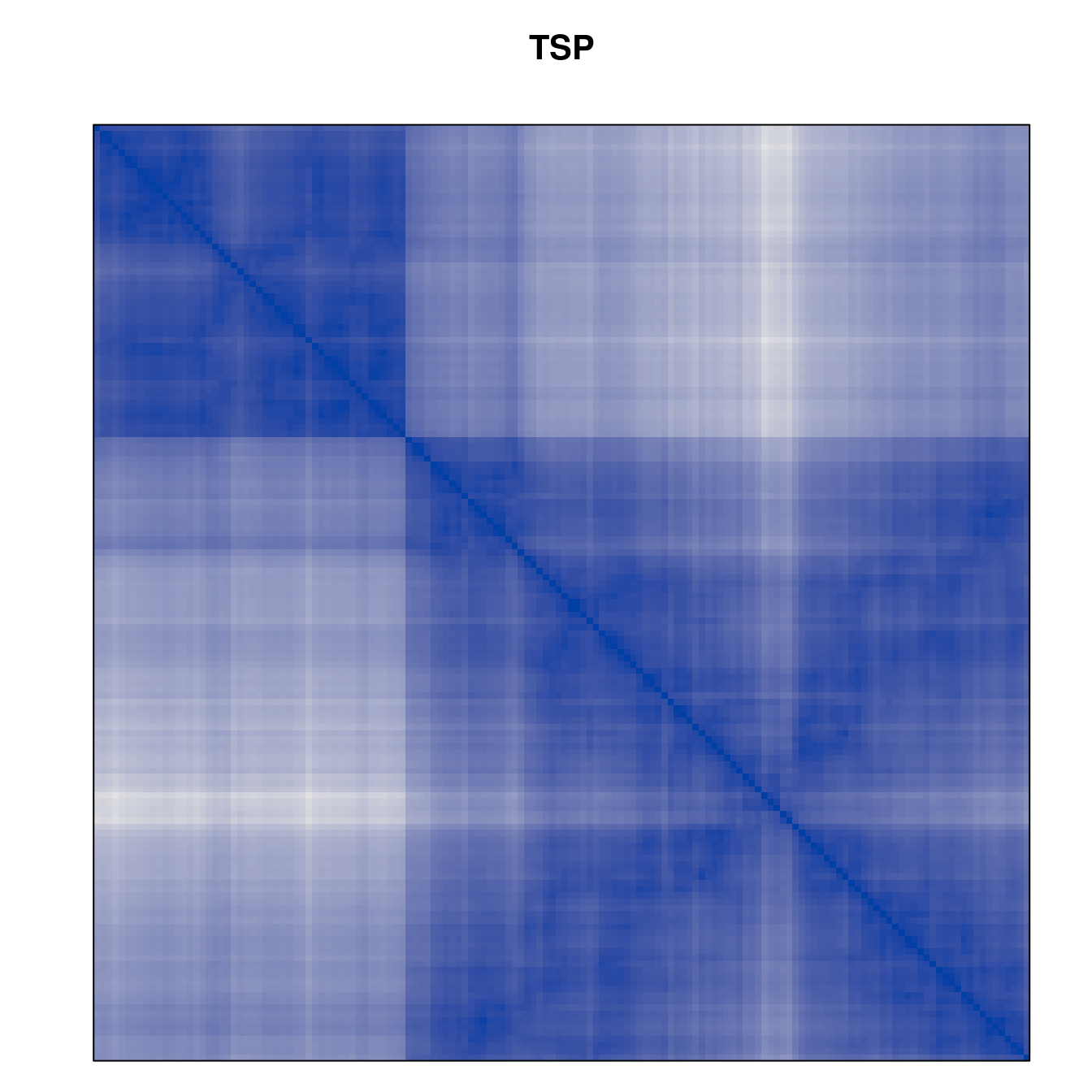
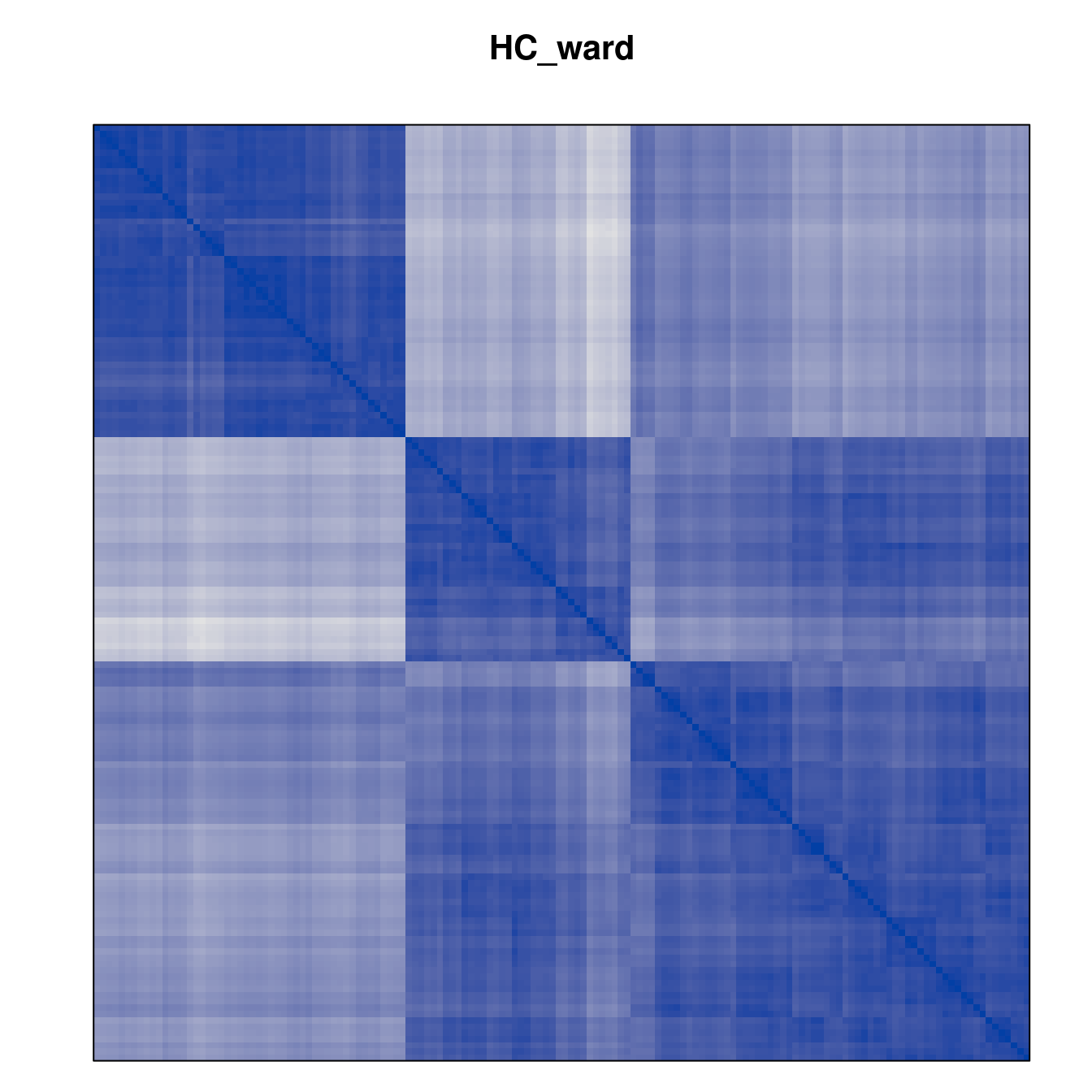
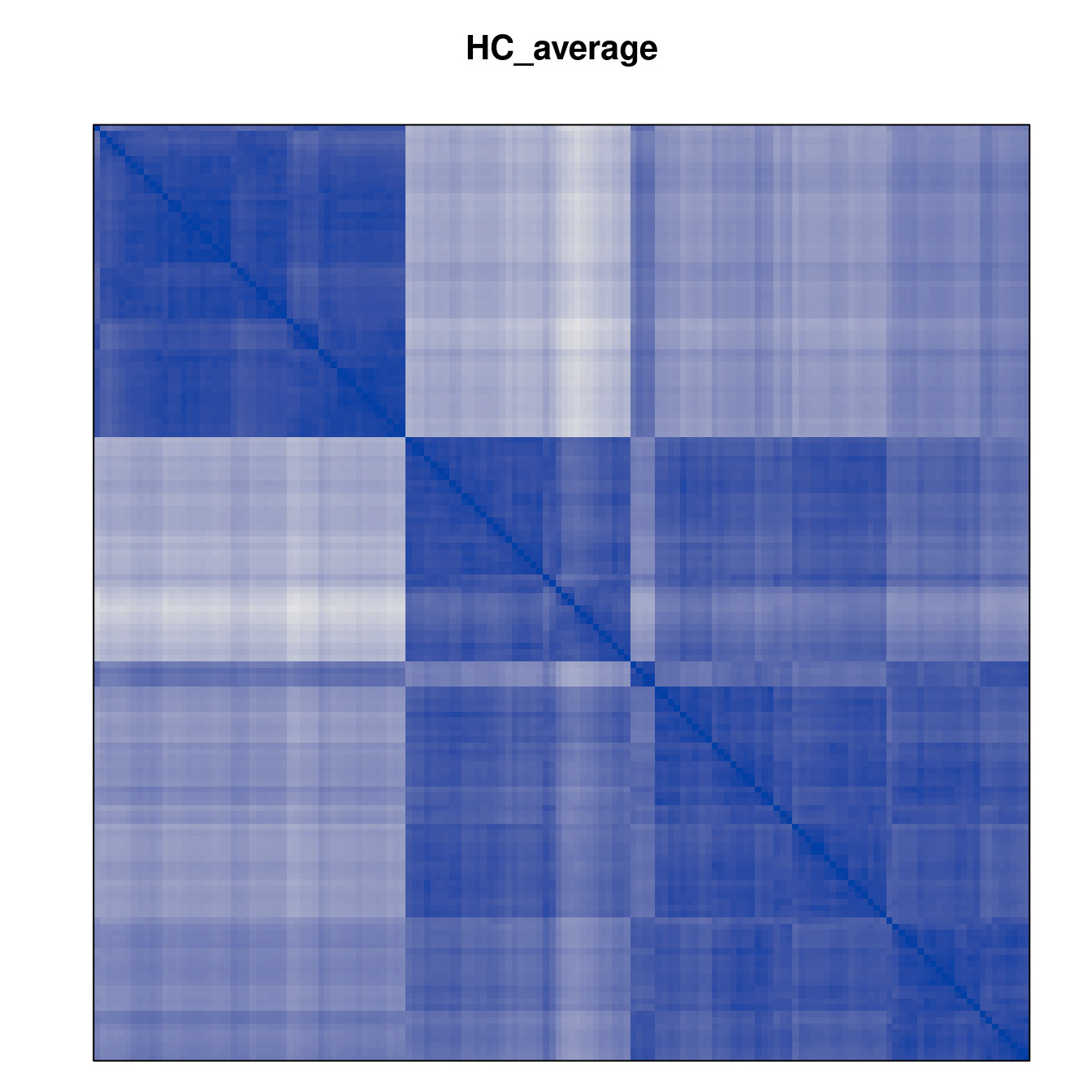
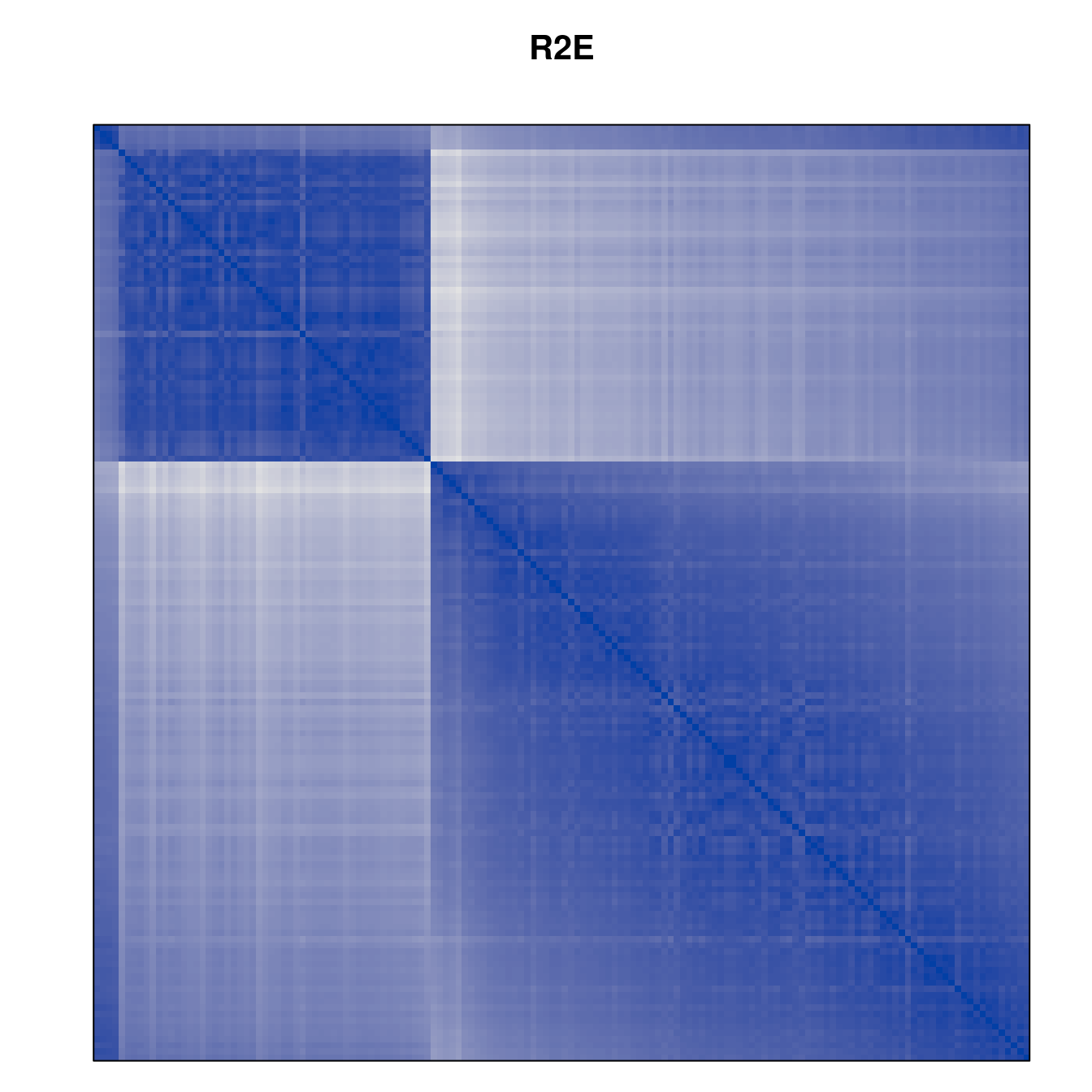
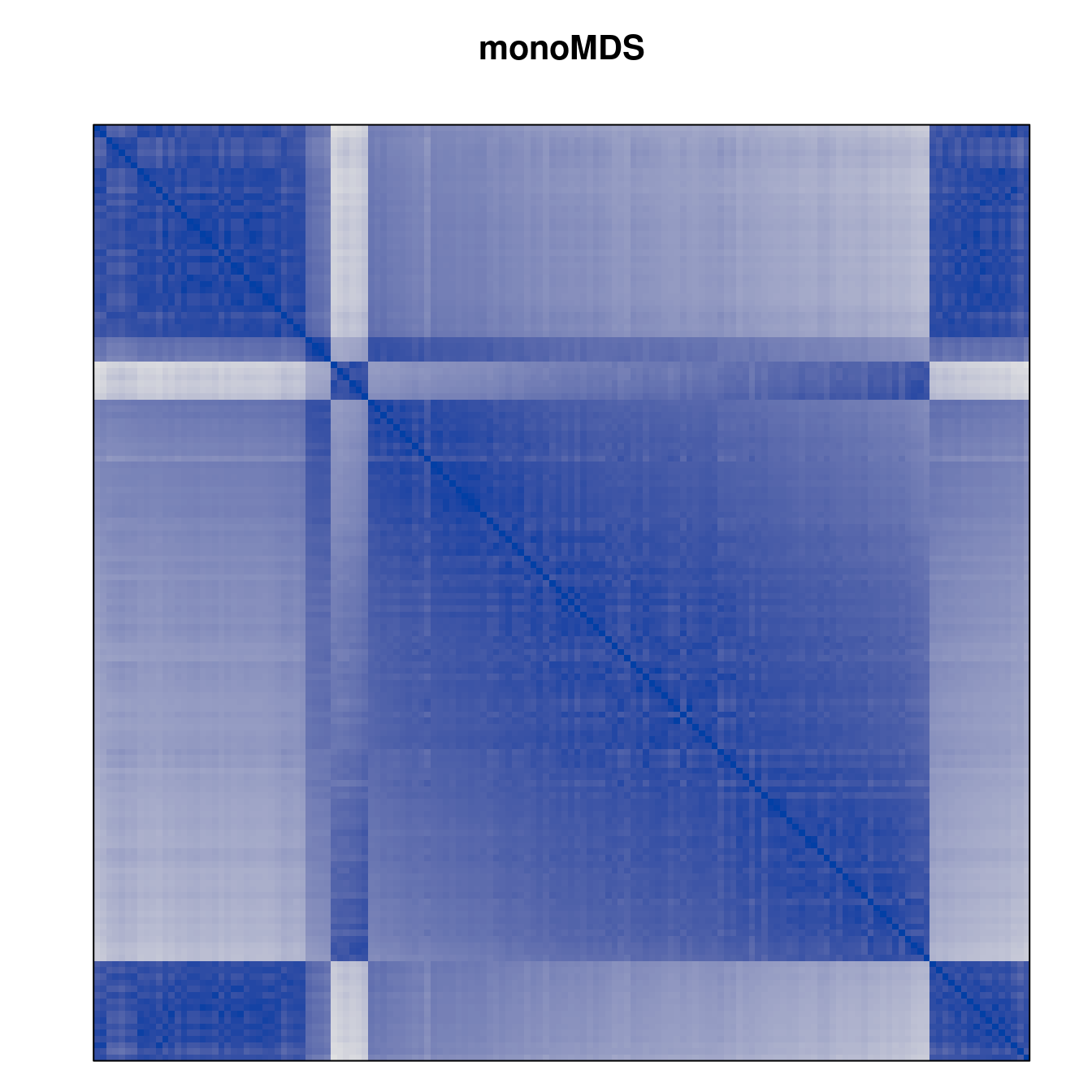



Comparison between methods
We can compare the seriation methods by how similar the orders are that they produce. The following code calculates distances between orders and then performs hierarchical clustering.
dst <- ser_dist(orders)
hmap(dst)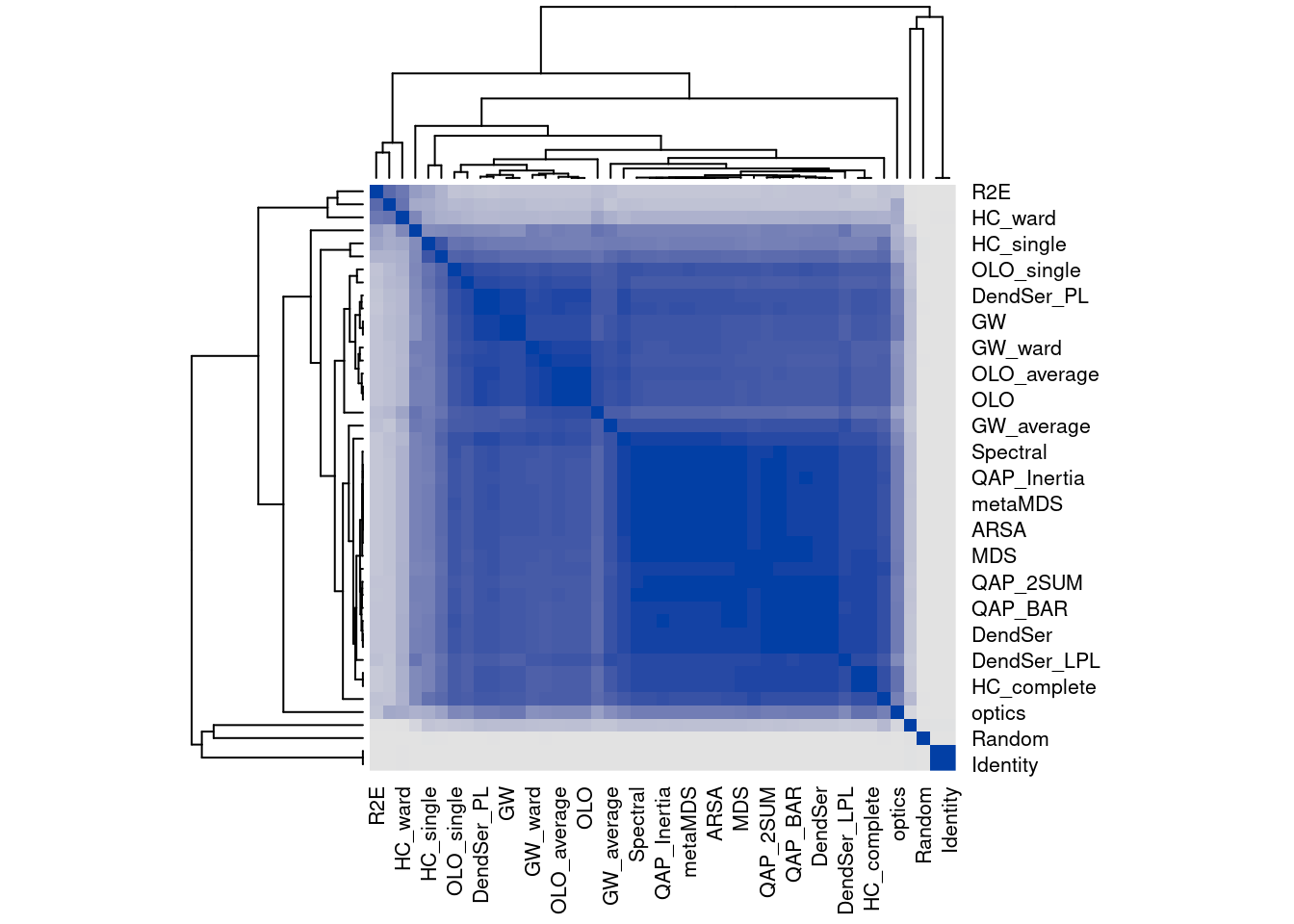
Here are the criterion measures. Some are maximized and some should be minimized. Details about the measures can be found in the manual page for criterion().
DT::datatable(round(criterion, 2))Matrix seriation
Matrix seriation reorders rows and columns of a data matrix. We perform the same steps as for distances in the previous section.
Methods
methods <- sort(list_seriation_methods("matrix"))
methods [1] "BEA" "BEA_TSP" "CA" "Heatmap" "Identity" "LLE"
[7] "Mean" "PCA" "PCA_angle" "Random" "Reverse" Performing seriation
orders <- list()
criterion <- list()
for (m in methods) {
cat(m)
tm <- system.time(orders[[m]] <- seriate(x, method = m))
criterion[[m]] <- data.frame(time = tm[1]+tm[2], rbind(criterion(x, orders[[m]])))
cat(" took", tm[1]+tm[2], "sec.\n")
}BEA took 0.122 sec.
BEA_TSP took 0.343 sec.
CA took 0.117 sec.
Heatmap took 0.339 sec.
Identity took 0.113 sec.
LLE took 0.142 sec.
Mean took 0.113 sec.
PCA took 0.113 sec.
PCA_angle took 0.114 sec.
Random took 0.114 sec.
Reverse took 0.111 sec.criterion <- do.call(rbind, criterion)Visualize the results
best_to_worse <- order(criterion[["Moore_stress"]], decreasing = FALSE)
orders <- orders[best_to_worse]
criterion <- criterion[best_to_worse, ]for (n in names(orders))
pimage(x, orders[[n]], main = n , key = FALSE)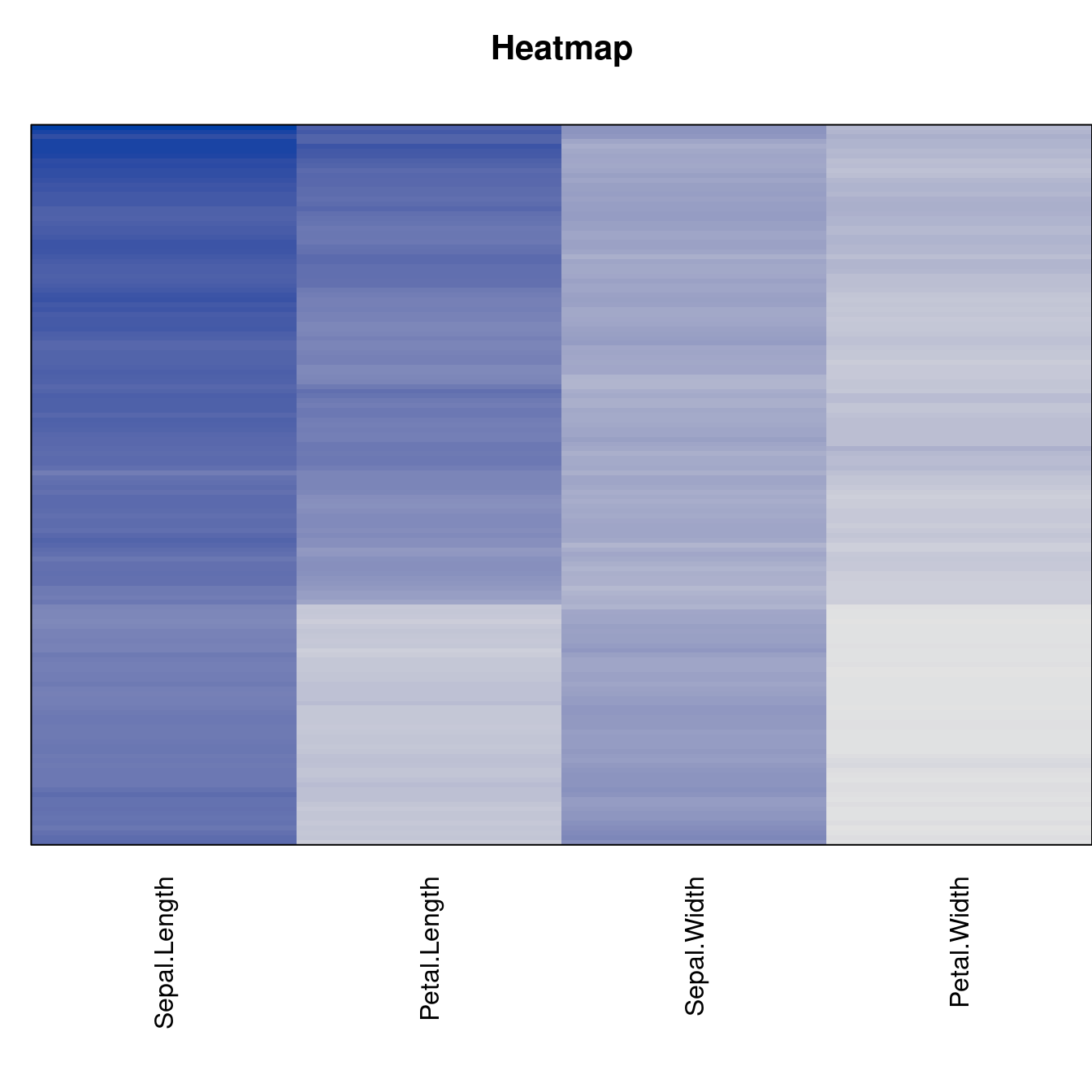
Comparison beween methods
DT::datatable(round(criterion, 2))